BAB II DASAR TEORI
advertisement

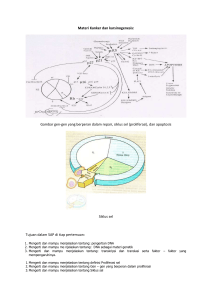
BAB II DASAR TEORI Pada bagian ini dijelaskan mengenai teori-teori yang mendukung pengelompokan data ekspresi gen, bentuk data ekspresi gen dan jenis analisis dari data ekspresi gen tersebut. Dasar-dasar teori ini digunakan untuk menyelesaikan tugas akhir. Dasar teori ini didapat dari studi literatur. 2.1 DNA (Deoxy-Ribonucleic Acid) Sel adalah bagian terkecil dari makhluk hidup. Setiap sel merupakan suatu sistem kompleks yang terdiri dari berbagai macam struktur pembangun yang dibungkus oleh membran [BRA01]. Pada setiap sel ini berbagai aktifitas sel, seperti metabolisme, pembelahan sel, ekspresi gen dilakukan secara teratur dan terkontrol. Pada umumnya makhluk hidup terbagi menjadi dua macam, prokariota dan eukariota, sehingga terdapat dua macam sel sesuai jenisnya. Perbedaan mendasar terletak pada ukuran dan struktur penyusun tubuhnya. Sel prokariota pada umumnya lebih kecil dan memiliki struktur yang lebih sederhana dibandingkan sel eukariota, misalnya: sel prokariota tidak memiliki membran dalam sel. Sel eukariota memiliki nukleus atau inti sel yang dipisahkan dari bagian sel lainnya dengan membran dalam. Pada inti sel eukariota ini terdapat rangkaian DNA (Deoxy-Ribonucleic Acid) yang menyimpan rangkaian instruksi untuk mengatur berbagai aktifitas sel. DNA merupakan pembawa informasi utama di suatu sel [BRA01]. Rangkaian instruksi yang tersimpan didalamnya mengatur aktifitas sel seperti metabolisme, pembelahan sel, dan ekspresi gen. Rangkaian instruksi pada DNA juga akan diturunkan kepada setiap sel anak yang dihasilkan, sehingga pada suatu makhluk hidup, setiap sel memiliki rangkaian DNA yang sama. DNA tersusun dari molekul kecil yang bernama nukleotida. Terdapat empat macam nukleotida yang berbeda yaitu: Adenosin, Guanien, Cytosine dan Thymine yang dilambangkan dengan A, C, G, dan T. Nukelotida ini membentuk suatu rangkaian DNA yang panjang dan memiliki struktur double helix. II-1 II-2 Rangkaian DNA tersebut memiliki kode-kode yang merepresentasikan ciri fisik makhluk hidup, bagaimana sel harus bekerja dan sebagainya. Pada dasarnya DNA akan mengalami transkripsi menjadi RNA (Ribo-nucleic Acid), yaitu rangkaian nukleotida mirip DNA dimana nukleotida Thymine diganti dengan Uracil (U). Pada proses transkripsi, molekul DNA pada salah satu benang disalin menjadi pre mRNA. Proses selanjutnya adalah splicing, yaitu membuang potongan rangkaian DNA yang tidak perlu (introns) dan menyambung semua potongan rangkaian yang akan dikodekan (exons) menjadi satu rangkaian mRNA. Setelah ini, dilakukan proses translasi, yaitu proses pembentukan protein dengan menyatukan asam amino yang terkodekan dengan urut pada mRNA. Asam amino dikodekan dengan tiga nukleotida yang urut pada DNA (disebut dengan triplet). Setiap triplet disebut dengan codon dan memiliki arti sebuah asam amino. Protein yang telah dibentuk memiliki fungsi yang berbeda-beda yang berkaitan dengan fungsi sel itu sendiri. Proses pengubahan DNA menjadi protein dapat dilihat pada gambar II-1. Gambar II-1 Proses perubahan DNA menjadi protein [BRA01] 2.2 Data Ekspresi Gen Ekspresi gen merupakan proses biologi dimana sekuen DNA diterjemahkan menghasilkan protein. Seiring perkembangan teknologi, telah ditemukan metode untuk menemukan urutan rangkaian DNA secara lengkap pada suatu makhluk hidup. Selain itu, perkembangan teknologi juga membawa penemuan teknologi cDNA dan microarray yang dapat mengukur tingkat ekspresi gen dalam skala besar. Microarray adalah perkembangan teknologi terakhir dalam biologi molekul, yang dapat mengukur tingkat ekspresi gen dari puluhan ribu gen secara paralel dan menghasilkan data yang sangat besar dan berharga [BRA00]. Dengan adanya data sebesar ini maka masalah utama yang muncul adalah menganalisis dan menangani data tersebut. II-3 Gambar II-2 Contoh microarray [AND06] Microarray pada dasarnya adalah suatu representasi pada sebuah bidang kaca (atau material lainnya), dimana molekul DNA diikatkan pada titik (spot) tertentu. Terdapat puluhan ribu titik pada sebuah array, masing-masing mengandung sejumlah besar molekul DNA atau fragmen dari molekul yang identik, yang panjangnya berkisar antara puluhan hingga ratusan nukloetida. Contoh cuplikan microarray dapat dilihat pada gambar II-2, dimana baris merepresentasikan gen dari suatu organisme dan kolom merepresentasikan sampel. Data mentah pada eksperimen microarray berbentuk gambar. Untuk mendapatkan data mengenai tingkat ekspresi gen, gambar tersebut harus dianalisis, masing-masing titik diidentifikasi, diukur intensitasnya dan dibandingkan dengan latar belakangnya. Data tersebut kemudian dimasukkan kedalam sebuah hasil akhir berupa matriks ekspresi gen, matriks inilah yang nantinya dianalisis untuk proses lebih lanjut. Pada matriks ini, baris merepresentasikan gen, dan kolom merepresentasikan berbagai macam sampel seperti tisu atau kondisi eksperimen. Angka pada baris dan kolom II-4 yang bersesuaian merepresentasikan tingkat ekspresi gen tertentu pada sampel tertentu. Tabel II-1 Matriks Ekspresi Gen Saccharomyces cerevisiae [EIS05] YORF YHR051W YKL181W YHR124W YHL020C YGR072W … Cell-cycle Alpha-Factor 1 0.03 0.33 0.36 -0.01 0.2 … Cell-cycle Alpha-Factor 2 0.3 -0.2 0.08 -0.03 -0.43 … … … … … … … … Contoh matriks ekspresi gen dapat dilihat pada tabel II-1. Matriks tersebut diambil dari organisme yeast (Saccharomyces cerevisiae). Baris pertama merupakan nama sampel pada kolom tersebut dan kolom pertama merupakan nama gen pada baris tersebut. Selain baris dan kolom yang telah disebutkan berisi tingkat ekspresi gen pada sampel tertentu (kolom) dan gen yang bersangkutan (baris). Contoh matriks ekspresi gen ini telah diperkecil, ukuran aslinya mencapai kurang lebih 6000 gen dan 80 sampel. 2.3 Analisis Data Ekspresi Gen Terdapat dua macam sisi bagaimana data ekspresi gen dianalisis [BRA00]: 1. membandingkan tingkat ekspresi dari masing-masing gen dengan membandingkan baris pada matriks ekspresi gen. 2. membandingkan tingkat ekspresi gen dari masing-masing sampel dengan membandingkan kolom pada matriks ekspresi gen. Dua metode di atas dapat dikombinasikan jika data sudah dinormalkan terlebih dahulu. Pada saat membandingkan baris ataupun kolom, dapat dicari kesamaan maupun perbedaan dari data yang ada. Misalnya, jika ditemukan bahwa ada dua baris yang mirip, maka dapat disimpulkan bahwa kedua gen yang bersangkutan mungkin memiliki fungsi yang mirip dalam sel. Jika yang dibandingkan adalah kolom, dapat dilihat gen mana yang dipengaruhi oleh kondisi sampel tertentu. II-5 Sebelum data dibandingkan, perlu ditemukan cara untuk menghitung kedekatan atau jarak dari dua buah objek yang dibandingkan. Objek – objek tersebut dapat dianggap sebagai sebuah titik pada dimensi n, atau sebuah vektor berdimensi n, dimana n adalah jumlah sampel untuk perbandingan gen, atau jumlah gen untuk perbandingan sampel. Metode yang biasa dipakai adalah penghitungan dengan jarak Euclidean. Jarak Euclidean dihitung dengan mencari akar dari jumlah kuadrat selisih masingmasing dimensi dari dua buah titik. Jika dituliskan sebagai rumus adalah sebagai berikut: d E ( x, y ) = n ∑ (x − y ) i =1 i 2 i Metode ini cukup valid untuk dipakai sebagai perhitungan kesamaan atau jarak dua buah objek yang dibandingkan pada matriks ekspresi gen. Meskipun menurut Alvis Brazma dan Jaak Vilo, tidak ada metode penghitungan jarak yang benar-benar valid [BRA00]. Gambar II-3 Analisis unsupervised (kiri) dan supervised (kanan) [BRA00] Setelah memilih metode penghitungan jarak yang sesuai, data ekspresi gen dapat dianalisis dengan cara supervised atau unsupervised. Pendekatan supervised dilakukan jika untuk beberapa atau semua data, ada informasi tambahan berupa fungsi utama dari gen atau kondisi sakit atau normal pada sampel yang ada. Informasi tambahan ini dapat dimasukkan pada matriks sebagai baris atau kolom tambahan. Dengan adanya informasi ini, tujuan utama analisis adalah membangun sebuah penggolong (classifier) yang mampu memprediksi informasi tambahan dari data baru yang diberikan. Sedangkan tujuan utama analisis unsupervised adalah mengelompokkan data untuk menemukan gen – gen atau sampel – sampel yang saling II-6 berkaitan. Sebagai perbandingan kedua analisis dapat dilihat pada gambar II-3. Misalkan data ekspresi gen digambarkan pada ruang berdimensi 2. Pada gambar di sebelah kiri, titik-titik yang memiliki kemiripan dicoba untuk dikelompokkan, sebagai contoh terdapat 3 cluster pada gambar, masing-masing terdiri dari beberapa titik yang saling berdekatan. Sebuah algoritma untuk analisis unsupervised harus dapat menemukan cluster tersebut. Pada gambar di sebelah kanan, sebagai contoh terdapat titik berisi dan titik berlobang, tujuan dari analisis unsupervised adalah menemukan sebuah aturan untuk menggolongkan titik-titik setepat mungkin. Sebagai contoh, garis putus-putus merupakan garis pemisah antara titik berisi dan titik berlobang. Selanjutnya dibahas satu persatu mengenai analisis supervised dan unsupervised. 2.3.1 Unsupervised Analysis Tujuan utama dari analisis ini adalah mengelompokkan (clustering) objek yang memiliki kesamaan. Pada umumnya, clustering dibagi menjadi 4 model [BRY05]: 1. Exclusive clustering Pada clustering ini, suatu objek hanya termasuk pada satu cluster saja. 2. Overlapping clustering Model clustering ini dapat memasukkan suatu objek pada beberapa cluster sekaligus. 3. Probabilistic clustering Suatu objek pada model clustering ini termasuk pada masing-masing cluster dengan probabilitas tertentu. 4. Hierarchical clustering Pada model clustering ini, semua objek secara kasar dibagi menjadi cluster pada tingkat tertinggi. Untuk setiap cluster, dilakukan pembagian lagi untuk level selanjutnya, hal ini dilakukan hingga cluster beranggotakan sebuah objek saja. Clustering bukan merupakan teknik baru, sudah banyak algoritma dikembangkan dan banyak yang sudah menerapkan algoritma tersebut untuk analisis data ekspresi gen. Untuk clustering data ekspresi gen, model yang umum digunakan adalah exclusive clustering dan hierarchical clustering. Algoritma yang telah digunakan antara lain: II-7 hierarchical, K-means dan self-organizing maps. Seperti pada paper DeRisi, yang menggunakan DNA untuk mempelajari metabolisme yeast. Beberapa penelitian lain dilakukan oleh Brazma, dan Van Helden untuk mempelajari regulasi sel [BRA00]. Algoritma hierarchical berjalan secara iteratif dengan menggabungkan dua cluster terdekat dimulai dengan cluster beranggotakan satu objek. Setelah menggabungkan dua cluster, jarak antara semua cluster yang ada dengan cluster yang baru dihitung ulang. Perlu dicatat bahwa untuk clustering yang lebih baik, perlu ditambahkan batas jarak yang ideal antar cluster yang merupakan masukan dari pengguna. Algoritma II-1 Algoritma K-means [HOO04] 1. Choose k initial center points randomly 2. Cluster data using Euclidean distance (or other distance metric) 3. Calculate new center points for each cluster using only points within the cluster 4. Re-Cluster all data using the new center points 1. This step could cause data points to be placed in a different cluster 5. Repeat steps 3 & 4 until the center points have moved such that in step 4 no data points are moved from one cluster to another or some other convergence criteria is met Algoritma pengelompokan K-means, dapat dilihat pada algoritma II-1, secara khusus menggunakan metode jarak Euclidean untuk menghitung jarak dua objek pada ruang berdimensi n. Pada awalnya, pengguna menentukan berapa banyaknya cluster yang diinginkan. Untuk menentukan jumlah cluster, belum ada teori atau algoritma yang baku [BRY05]. Setelah itu ruang dimensi dibagi menjadi sejumlah cluster sesuai masukan pengguna. Algoritma berjalan secara iteratif dengan menghitung ulang titik tengah masing-masing cluster dan menyesuaikan diri dengan setiap objek yang dimasukkan ke cluster terdekat. Proses ini terus berjalan hingga mencapai keadaan stabil atau batas maksimal iterasi terlampaui. Penentuan cluster awal sebelum algoritma berjalan dapat bermacam-macam, salah satunya ditentukan secara acak. II-8 Analisis unsupervised ini telah digunakan untuk clustering gen maupun sampel. Proses clustering gen untuk menemukan gen-gen yang saling berkaitan fungsinya bisa dilakukan jika terdapat sekumpulan data gen lengkap dari suatu organisme. Beberapa penelitian telah dilakukan untuk menemukan gen-gen yang saling berkaitan dalam fungsi metabolisme sel [BRA00]. Sedangkan untuk mengelompokkan sampel tidak diperlukan data gen lengkap dari suatu organisme, karena dengan hanya sedikit gen dapat dicari nilai kedekatan atau kesamaan dari dua buah sampel. Cluster yang telah dihasilkan dapat diperiksa keabsahannya melalui beberapa cara [HOO04]: 1. ukuran (diameter) cluster dibandingkan dengan jarak antar cluster 2. jarak antara masing-masing anggota cluster dengan pusat cluster 3. diameter dari cluster terkecil 2.3.2 Supervised Analysis Tujuan utama dari analisis supervised dari data ekspresi gen adalah membentuk classifier seperti garis pemisah lanjar, pohon keputusan atau support vector machines (SVM) yang memetakan objek-objek ke suatu kelas tertentu. Sebagai contoh, jika sebuah classifier dapat dibangun dari sejumlah data yang dapat memisahkan sampel yang terkena tumor dan tidak, maka dapat dipakai untuk diagnosa tumor lebih dini. Lebih lanjut lagi, jika classifier tersebut berdasar pada aturan yang sederhana, dapat dipelajari mekanisme dalam pembentukan tumor. Pembentukan classifier didasari pada teori pembelajaran mesin. Sebuah classifier dilatih dengan sejumlah data latih yang sudah diketahui kelasnya. Kemudian, classifier tersebut, dapat berupa aturan atau yang lainnya, diuji dengan sejumlah data uji untuk memeriksa keabsahannya. Jika memenuhi kualitas ketelitian tertentu, classifier dapat digunakan untuk sejumlah data yang belum diketahui kelasnya. Perlu dicatat bahwa, untuk menggolongkan sampel, terdapat masalah dimana atribut data (gen) jauh lebih banyak daripada objek (sampel) yang digolongkan. Hal ini menyebabkan mudahnya menemukan pemisah yang sempurna jika tidak hati-hati dalam membatasi keruwetan classifier yang dibuat. Untuk menghindari masalah II-9 tersebut harus dicari classifier yang sangat sederhana, sambil tetap menjaga akurasi classifier. Kedua analisis di atas dapat digunakan bersama, dengan pada awalnya menggunakan analisis unsupervised untuk mencari cluster dengan keterkaitan tinggi. Kemudian, digunakan analisis supervised pada salah satu cluster saja untuk membatasi jumlah atribut yang terlalu banyak.