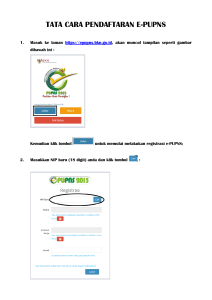
laporan praktek kerja profesi apoteker di pt
advertisement

UNIVERSITAS INDONESIA LAPORAN PRAKTEK KERJA PROFESI APOTEKER DI PT. NOVARTIS INDONESIA JL. PROF. DR. SATRIO KAV. 18 KUNINGAN CITY SETIABUDI JAKARTA SELATAN 12940 PERIODE 4 FEBRUARI - 28 MARET 2014 LAPORAN PRAKTEK KERJA PROFESI APOTEKER ASVINASTUTI RIKASIH, S. Farm. 1306343416 ANGKATAN LXXVIII FAKULTAS FARMASI PROGRAM PROFESI APOTEKER DEPOK JUNI 2014 Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 UNIVERSITAS INDONESIA LAPORAN PRAKTEK KERJA PROFESI APOTEKER DI PT. NOVARTIS INDONESIA JL. PROF. DR. SATRIO KAV. 18 KUNINGAN CITY SETIABUDI JAKARTA SELATAN 12940 PERIODE 4 FEBRUARI - 28 MARET 2014 LAPORAN PRAKTEK KERJA PROFESI APOTEKER Diajukan sebagai salah satu syarat untuk memperoleh gelar Apoteker ASVINASTUTI RIKASIH, S. Farm. 1306343416 ANGKATAN LXXVIII FAKULTAS FARMASI PROGRAM PROFESI APOTEKER DEPOK JUNI 2014 ii Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 iii Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 iv Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 v Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 KATA PENGANTAR Puji syukur kehadirat Allah SWT yang senantiasa melimpahkan karunia dan rahmat-Nya sehingga penulis dapat menyelesaikan Laporan Praktek Kerja Profesi Apoteker (PKPA) di PT. Novartis Indonesia. Laporan Praktek Kerja Profesi Apoteker ini disusun sebagai salah satu syarat yang harus dipenuhi oleh mahasiswa Program Profesi Apoteker di Fakultas Farmasi Universitas Indonesia untuk mencapai gelar profesi Apoteker. Selain itu juga memberikan kesempatan kepada mahasiswa untuk memahami peran dan tugas Apoteker di Industri Farmasi, khususnya di PT. Novartis Indonesia bagian Drug Regulatory Affair (DRA). Pelaksanaan Praktek Kerja Profesi Apoteker (PKPA) di PT. Novartis Indonesia berlangsung pada periode 4 Februari – 28 Maret 2014. Penulis menyadari bahwa tanpa bantuan dari berbagai pihak, penulis tidak dapat menyelesaikan Laporan PKPA ini. Pada kesempatan ini, penulis ingin menyampaikan terima kasih atas bantuan dan bimbingan kepada: 1. Halimah Lailasari, S.Farm., Apt selaku pembimbing di PT. Novartis Indonesia yang telah banyak memberikan bimbingan dan arahan kepada penulis selama melaksanakan PKPA. 2. Dr. Hasan Rachmat, DEA., Apt., selaku pembimbing di Program Profesi Apoteker Fakultas Farmasi UI. 3. Bapak Firnando Sianturi, selaku Head of DRA PT. Novartis Indonesia 4. Ibu Hasriani Yusuf, selaku Chief Scientific Officer PT. Novartis Indonesia yang telah memberikan kesempatan kepada penulis untuk melakukan PKPA di PT. Novartis Indonesia. 5. Bapak Dr. Mahdi Jufri, M.Si. selaku Dekan Fakultas Farmasi UI 6. Bapak Dr. Hayun, M. Si., selaku Ketua Program Profesi Apoteker Fakultas Farmasi Universitas Indonesia. 7. Bapak Dr. Harmita, Apt., selaku Pembimbing akademis Program Profesi Apoteker Fakultas Farmasi Universitas Indonesia. 8. Bapak dan Ibu staf pengajar beserta segenap karyawan Program Profesi Apoteker Fakultas Farmasi Universitas Indonesia. vi Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 9. Seluruh staf di PT. Novartis Indonesia, khususnya Para Staf dan Manajer bagian Drug Regulatori Affair PT. Novartis Indonesia. 10. Seluruh keluarga atas kasih sayang, perhatian, kesabaran, dorongan semangat, doa, dan dukungan baik moral maupun finansial yang selama ini diberikan kepada penulis. 11. Teman-teman angkatan LXXVIII Program Profesi Apoteker Universitas Indonesia dan pihak-pihak lain yang tidak dapat disebutkan satu per satu yang telah membantu baik langsung maupun tidak langsung dalam pembuatan laporan ini. Penulis berharap Allah SWT berkenan membalas segala kebaikan semua pihak yang telah membantu. Penulis menyadari bahwa laporan ini masih jauh dari kesempurnaan. Oleh karena itu, penulis dengan senang hati menerima segala kritik dan saran yang membangun demi tercapainya hasil yang lebih baik lagi dari pembaca. Penulis berharap bahwa laporan ini dapat memberikan manfaat bagi para pembaca dengan memberikan pengetahuan dan pengalaman yang penulis peroleh dari Praktek Kerja Profesi Apoteker. Penulis 2014 vii Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 ABSTRAK Nama : Asvinastuti Rikasih Program Studi : Apoteker Judul : Laporan Praktek Kerja Profesi Apoteker di PT. Novartis Indonesia Jl. Prof. Dr. Satrio Kav. 18 Kuningan City Setiabudi Jakarta Selatan 12940 Periode 4 Februari - 28 Maret 2014 Praktik Kerja Profesi Apoteker di PT. Novartis Indonesia bertujuan agar memahami tugas Apoteker, khususnya di bagian Drug Regulatory Affairs dalam proses meregistrasikan obat baru, registrasi variasi pada produk yang telah memiliki Nomor Izin Edar, dan meregistrasikan ulang produk yang telah habis masa izin edarnya. Registrasi adalah prosedur pendaftaran dan evaluasi suatu produk untuk mendapatkan izin edar. Tugas khusus yang diberikan adalah penyusunan dan proses pengajuan dokumen registrasi variasi terkait penandaan produk B PT. Novartis Indonesia ke Badan POM. Tahapan yang dilakukan yaitu adanya informasi perubahan dari Novartis global ke DRA Novartis Indonesia, pengambilan dokumen dari sistem informasi terkait, penyusun dossier dan pengecekan kelengkapan dokumen, dan proses submit atau pengajuan dokumen di BPOM. Dokumen yang diserahkan ke BPOM untuk registrasi variasi mengikuti ketentuan ACTD dan penyusunan serta pengajuannya berdasarkan pada keputusan Kepala BPOM Republik Indonesia Nomor HK.03.1.23.10.11.08481 Tahun 2011 Tentang Kriteria dan Tata Laksana Registrasi Obat. Kata kunci Tugas umum Daftar acuan Tugas khusus Daftar acuan : izin edar, Novartis Indonesia, penandaan, produk B, registrasi : xii + 11 halaman; 7 lampiran : 3 (2010-2014) : vi + 31 halaman; 2 gambar; 1 tabel; 7 lampiran : 8 (2008-2014) viii Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia ABSTRACT Name : Asvinastuti Rikasih Program Study : Apothecary Title : Report of Apothecary Profession Internship at PT. Novartis Indonesia Jl. Prof. Dr. Satrio Kav. 18 Kuningan City Setiabudi Jakarta Selatan 12940 on February 4th - March 28th 2014 Pharmacists Professional Practice at PT. Novartis Indonesia aims to understand the Pharmacists duties in drug industry, particularly in the Drug Regulatory Affairs in the process of registering new drugs, registration of variations on a product that has had a Marketing Authorization Number, and re-register the product with an expired license orbit. Registration is the procedure to registry and evaluate product to obtain marketing authorization. Specific assignment has given titled the preparation and submission process of variation registration documents related labeling of Product B PT. Novartis Indonesia to the BPOM. Steps taken by received information changes from the Novartis global to DRA Novartis Indonesia, document retrieval from related information systems, compilers dossier and checking the documents, and the submission process of documents in the BPOM. Documents submitted to the BPOM for registration variations follow the provisions of ACTD and the preparation and submission is based on keputusan Kepala BPOM Republik Indonesia Nomor HK.03.1.23.10.11.08481 Tahun 2011 Tentang Kriteria dan Tata Laksana Registrasi Obat. Keywords : labeling, marketing authorization, Novartis Indonesia, product B, registration General Assignment : xii + 11 pages; 7 appendices Bibliography : 3 (2010-2014) Specific Assignment : vi + 31 pages; 2 pictures; 1 tables; 7 appendices Bibliography : 8 (2008-2014) ix Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia DAFTAR ISI HALAMAN JUDUL .......................................................................................... ii HALAMAN PERNYATAAN ORISINALITAS ................................................. iii SURAT PERNYATAAN BEBAS PLAGIARISME ............................................ iv HALAMAN PENGESAHAN .............................................................................. v KATA PENGANTAR ......................................................................................... vi HALAMAN PERSETUJUAN PUBLIKASI KARYA ILMIAH .......................... viii ABSTRAK ......................................................................................................... ix ABSTRACT ....................................................................................................... x DAFTAR ISI ....................................................................................................... xi DAFTAR LAMPIRAN ........................................................................................ xii BAB 1. PENDAHULUAN ................................................................................. 1.1. Latar Belakang ................................................................................. 1.2. Tujuan .............................................................................................. 1 1 3 BAB 2. TINJAUAN UMUM ............................................................................. 2.1. PT. Novartis Indonesia ..................................................................... 2.1.1. Sejarah dan Perkembangan ...................................................... 2.1.2. Visi dan Misi .......................................................................... 2.1.3. Kewargaan Perusahaan .......................................................... 2.2. Struktur Organisasi PT. Novartis Indonesia ...................................... 2.2.1. Struktur Organisasi Head Office PT. Novartis Indonesia .......... 2.2.2. Struktur Organisasi Medical and Regulatory Department ......... 4 4 4 5 5 7 8 8 BAB 3. TINJAUAN KHUSUS .......................................................................... 13 3.1. Kriteria dan Tata Laksana Registrasi Obat ........................................ 13 3.2. ASEAN Common Technical Dossier/ ASEAN Common Technical Requirements .................................................................................. 21 BAB 4. PEMBAHASAN .................................................................................... 4.1. Registrasi Obat .................................................................................. 4.2. Registrasi Obat Baru ......................................................................... 4.3. Registrasi Obat Copy ........................................................................ 4.4. Registrasi Variasi .............................................................................. 4.5. Registrasi Ulang ............................................................................... 4.6. Regulator .......................................................................................... 4.6.1. Fungsi Regulator ................................................................... 4.6.2. Karakterisasi Regulator ......................................................... 24 24 25 27 28 28 29 29 29 BAB 5. KESIMPULAN DAN SARAN .............................................................. 30 5.1. Kesimpulan ...................................................................................... 30 5.2. Saran ................................................................................................. 30 DAFTAR ACUAN ............................................................................................ 31 x Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 DAFTAR LAMPIRAN Lampiran 1 Struktur organisasi Medical and Regulatory Departement PT. Novartis Indonesia .......................................................................... Lampiran 2 Formulir registrasi ........................................................................... Lampiran 3 Isi dokumen pra-registrasi dan registrasi .......................................... Lampiran 4 Alur proses pra-registrasi ................................................................. Lampiran 5 Alur proses registrasi........................................................................ Lampiran 6 Dokumen registrasi baru ................................................................. Lampiran 7 Dokumen registrasi variasi .............................................................. xi Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 33 34 38 43 44 45 52 Universitas Indonesia 1 BAB 1 PENDAHULUAN 1.1 Latar Belakang Salah satu komponen kesehatan yang sangat penting adalah tersedianya obat sebagai bagian dari pelayanan kesehatan masyarakat. Obat memiliki berbagai fungsi, yaitu untuk diagnosis, pencegahan, penyembuhan, pemulihan, atau peningkat kesehatan. Obat yang digunakan harus memenuhi standard mutu yang dipersyaratkan, yaitu harus memenuhi persyaratan keamanan pemakaian (safety), persyaratan mutu kegunaan (efficacy), dan persyaratan kualitas produk (quality). Industri farmasi adalah badan usaha yang memiliki izin dari Menteri Kesehatan untuk melakukan kegiatan pembuatan obat atau bahan obat menurut Peraturan Menteri Kesehatan RI No. 1700/MenKes/Per/XII/2010. Tingginya kebutuhan akan obat dalam dunia kesehatan dan vitalnya aktivitas obat mempengaruhi fungsi fisiologi tubuh manusia melahirkan sebuah tuntutan terhadap industri farmasi agar mampu memproduksi obat yang berkualitas (Keputusan Menteri Kesehatan RI, 2010). Salah satu upaya yang dilakukan industri farmasi dalam rangka meningkatkan kualitas obat yang diproduksinya yaitu dengan menerapkan CPOB (Cara Pembuatan Obat yang Baik) yang sifatnya dinamis, sesuai dengan perkembangan zaman. Melalui pedoman CPOB, semua aspek yang berhubungan dengan produksi dan pengendalian mutu obat diperhatikan dan ditentukan sedemikian rupa dengan tujuan untuk menjamin bahwa produk obat dibuat senantiasa memenuhi persyaratan mutu yang telah ditentukan sesuai dengan tujuan penggunaannya. Meningkatnya pertumbuhan jumlah dan jenis produk yang beredar baik lokal maupun impor, menyebabkan penggunaan produk obat di masyarakat juga meningkat, tetapi keadaan ini tidak selalu diikuti oleh pengetahuan yang memadai untuk memilih dan menggunakan produk secara tepat dan aman. Dikhawatirkan persaingan bisnis hanya akan mengedepankan profit tanpa memperhatikan faktor manfaat dan keamanannya, sehingga dapat beresiko pada kesehatan dan keselamatan masyarakat sebagai konsumen. Oleh karena itu, selain kewajiban dalam penerapan CPOB bagi setiap industri farmasi, pemerintah juga membentuk 1 Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia 2 suatu badan yang disebut Badan Pengawas Obat dan Makanan (Badan POM) yang memiliki jaringan nasional dan koordinasi internasional serta kewenangan penegakan hukum dan memiliki kredibilitas profesional yang tinggi. Sebagai institusi pemerintah yang memiliki kewenangan dan tanggung jawab dalam pengawasan produk obat dan makanan dengan membuat ketentuan tentang pendaftaran produk, baik obat sintesis, produk biologi, obat tradisional, obat herbal, dan fitofarmaka, pangan maupun suplemen makanan dari produsen baik dalam maupun luar negeri, BPOM mengendalikan peredaran dan juga pengawasan dengan cara mengadakan proses registrasi yang harus dilalui setiap industri farmasi dalam proses pemasaran produknya. Dalam pelaksanaan pendaftaran atau registrasi obat, terdapat suatu prosedur dimana diperlukan bagian yang mengerti dan memahami secara jelas mengenai tata laksana prosedur registrasi tersebut, terkait persyaratan dan peraturanperaturan pemerintah terkait. Oleh karena itu, masing-masing industri memiliki bagian yang disebut dengan Regulatory Affairs yang merupakan penghubung antara pihak industri dengan pihak pemerintah, dalam hal ini BPOM. Regulator yang bekerja di bagian Regulatory Affairs harus dapat menjamin bahwa produk yang akan diregistrasikan memiliki dokumen yang menyatakan bahwa produk telah memenuhi persyaratan mutu, keamanan, dan efikasi. Apoteker dalam peranannya sebagai salah satu tenaga kesehatan profesional, memiliki tanggung jawab tersebut dan merupakan satu-satunya profesi yang dapat melaksanakan tugas tersebut. Sebagai profesi yang memiliki pengetahuan tentang obat, apoteker dituntut untuk dapat meregistrasikan suatu produk obat. Pembekalan pengetahuan, keterampilan, dan pemahaman calon apoteker yang komprehensif antara teori dan praktek langsung sangat diperlukan. Pembekalan ini dapat memberikan gambaran kepada calon apoteker mengenai tanggung jawabnya di industri farmasi, khususnya di bagian Regulatory Affair sehingga dapat menjalankan perannya secara profesional saat bekerja di industri farmasi nantinya. Praktek Kerja Profesi Apoteker (PKPA) merupakan salah satu sarana bagi calon apoteker untuk mendapatkan pengalaman dan informasi yang lebih dalam mengenai tugas dan fungsi apoteker di industri farmasi tersebut. Oleh karena itu, Program Profesi Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 3 Apoteker Fakultas Farmasi Universitas Indonesia bekerja sama dengan Industri Farmasi PT. Novartis Indonesia dalam menyelenggarakan Praktek Kerja Profesi Apoteker (PKPA). Pelaksanaan PKPA ini berlangsung selama dua bulan, yaitu dari tanggal 4 Februari 2014 hingga 28 Maret 2014 di bagian Drug Regulatory Affair (DRA) PT. Novartis Indonesia. 1.1 Tujuan Praktik Kerja Profesi Apoteker (PKPA) di PT. Novartis Indonesia khususnya di Medical & Regulatory Department, bertujuan agar para calon Apoteker memahami tugas Apoteker di industri farmasi, khususnya di bagian Drug Regulatory Affairs di PT. Novartis Indonesia, Jakarta dalam proses meregistrasikan obat baru, registrasi variasi pada produk yang telah memiliki Nomor Izin Edar, dan meregistrasikan ulang produk yang telah habis masa izin edarnya. Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 4 BAB 2 TINJAUAN UMUM 2. 1 PT. Novartis Indonesia 2. 2. 1 Sejarah dan Perkembangan Sejarah Novartis di Indonesia berawal dengan berdirinya PT. Ciba Indonesia pada 1968. Setelah melalui beberapa momentum, mengikuti penggabungan global dua perusahaan, yaitu Sandoz dan Ciba Geigy menjadi Novartis, pada tahun 1996 PT. Ciba Indonesia bergabung dengan PT. Sandoz Biochemie Pharma membentuk PT. Novartis Biochemie. Pada tahun 2006, Novartis Global mengakuisisi Hexal Group, diikuti juga oleh akusisi PT. Prima Hexal di Indonesia oleh Divisi Sandoz Novartis Indonesia dan kemudian mendirikan badan hukum baru yakni PT. Sandoz Indonesia. Pada tahun 2006, tepat memasuki sepuluh tahun setelah merger global, PT Novartis Biochemie berubah nama menjadi PT. Novartis Indonesia (Novartis Indonesia, 2014). Dua anak perusahaan lainnya, yakni PT Ciba Vision dan PT. Sandoz Indonesia. PT Ciba Vision Batam sebagai pemasok utama lensa kontak di seluruh dunia dan memperkerjakan 3000 karyawan di Pulau Batam. Sedangkan PT. Sandoz Indonesia memproduksi obat-obat generik dan memiliki 600 pegawai yang beroperasi di Pasar Rebo, Jakarta Timur (Novartis Indonesia, 2014). Pada saat ini, Novartis mempekerjakan sekitar 650 tenaga kerja di Indonesia pada bidang Farmasi dan Produk Kesehatan. Perusahaan juga memiliki laboratorium penelitian NITD Hasanuddin Research Institut (NHCRI) yang memusatkan penelitiannya pada pengembangan obat-obat penyakit tropis seperti penyakit Deman Berdarah (DBD), Tuberkulosis (TBC), dan Malaria di Makassar, Sulawesi. Novartis memproduksi obat-obatan seperti Voltaren (natrium diklofenak), Cataflam (kalium diklofenak), Clozaril (clozapin), Tegretol (karbamazepin), Valsartan Ni (valsartan), Diovan (valsartan), dan lain-lain. Agen tambahan termasuk Sandimmun Neoral (siklosporin), Femara (letrozol), Ritalin (metilfenidat), Lamisil (terbinafin), dan lain-lain (Novartis Indonesia, 2014). 4 Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia 5 2. 1. 2 Visi dan Misi Novartis Indonesia Visi dari Novartis Indonesia adalah “Menjadi perusahaan yang paling dihormati oleh para pemangku kepentingan di industri farmasi”. Hal ini diwujudkan dalam misi-misi yang akan dilakukan oleh Novartis Indonesia, antara lain: a. Menjadi perusahaan farmasi nomor 1 dengan pertumbuhan positif pada tahun 2016. b. Memaksimalkan portfolio obat-obatan 'primary care' dan meningkatkan pertumbuhan obat-obatan 'speciality' yang inovatif c. Mempekerjakan dan mengembangkan talenta muda berpotensi, serta menciptakan lingkungan kerja yang sehat dan kondusif bagi karyawan untuk mengembangkan diri d. Meningkatkan kesehatan masyarakat Indonesia melalui ketersediaan dan keterjangkauan obat-obatan inovatif e. Bermitra dengan para pemangku kepentingan melalui berbagai kerjasama dan kolaborasi untuk menangani masalah-masalah kesehatan 2. 1. 3 Kewargaan Novartis Indonesia 2. 1. 3. 1 Akses ke Obat-obatan Novartis Indonesia juga menunjukkan komitmennya dengan mendukung komunitas lokal. Sejak 2003, Novartis Indonesia telah membantu para penderita Chronic Myeloid Leukemia (CML) untuk mendapatkan perawatan secara gratis bekerjasama dengan Yayasan Kanker Indonesia. Novartis Indonesia juga secara aktif berpartisipasi dalam penanganan bencana, antara lain membantu penyediaan perpustakaan keliling untuk Komisi Nasional Perlindungan Anak pasca tsunami 2005, dan bantuan spontan para karyawan secara sukarela pada bencana gempa Jogja tahun 2006 (Novartis Indonesia, 2014). Dengan visi untuk meningkatkan akses bagi para pasien untuk memperoleh obat-obatan, Novartis Indonesia secara berkesinambungan telah menjalin kemitraan dengan pemerintah melalui program ASKES dimana banyak obat-obatan terbaik Novartis termasuk dalam program asuransi pemerintah tersebut (Novartis Indonesia, 2014). Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 6 2. 1. 3. 2 Program Dalam rangka memperbesar organisasinya di Indonesia untuk dapat meningkatkan kontribusinya bagi masyarakat Indonesia, Novartis Indonesia meluncurkan sebuah program transformasi organisasi yaitu “SEHATi Bersama, Sehatkan Indonesia!”. Program yang dicanangkan pada tanggal 1 November 2011 ini diaktualiasikan dalam 3 sub program, yakni (Novartis Indonesia, 2014): a. “Sehati Berbakti” merupakan komitmen Novartis yang diwujudkan melalui dedikasi perusahaan dalam menciptakan dan memberikan inovasi obat-obatan baru untuk menyembuhkan, menyelamatkan dan meningkatkan kualitas pasien di Indonesia. Kehadiran Novartis selama lebih dari 30 tahun di Indonesia telah membuktikan dedikasinya dan hal ini akan terus dipertahaankan. b. “Sehati Bersinergi” merupakan bentuk semangat bersinergi dengan semua elemen dari para pengampu kepentingan dalam melakukan beberapa kegiatan, yaitu penelitian klinis yang akan dilakukan di Indonesia untuk menemukan obat baru. Dimana kegiatan ini akan membuka kesempatan alih pengetahuan bagi para peneliti dan pakar riset di Indonesia, serta kemitraan dengan pemerintah maupun pihak terkait untuk meningkatkan akses terhadap pengobatan inovatif baik yang sudah ada maupun yang baru c. “Sehati Berkinerja” merupakan perwujudan kinerja perusahaan yang lebih baik dan mengarah pada pengembangan bisnis dan organisasi di Indonesia. Dengan demikian Novartis mampu menyediakan lebih banyak kesempatan kerja bagi putra putri Indonesia serta melakukan pengembangan sumber daya manusia di bidang farmasi dan bioteknologi. 2. 1. 3. 3 Novartis Community Oartnership Day (NCPD) NCPD merupakan inisiatif tahunan yang dilakukan oleh Novartis. Inisiatif ini dilakukan secara global di setiap negara yang dimana Novartis beroperasi. Lebih dari 16.500 karyawan Novartis berpartisipasi dalam NCPD seluruh dunia. Di Indonesia lebih dari 350 karyawan Novartis dan Sandoz berpartisipasi dalam NCPD tahun 2013 ini. Melalui implementasi program NCPD tahun ini, kita memberikan kesempatan kepada karyawan Novartis untuk Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 7 mengkontribusikan waktu mereka untuk diberikan ke lingkungan dan juga orangorang yang tinggal di sekitar tempat mereka bekerja (Novartis Indonesia, 2014). NCPD juga merupakan komponen yang paling penting dalam budaya perusahaan sebagai realisasi dari komitmen Novartis untuk berkontribusi dan memberikan sesuatu yang bermanfaat bagi lingkungan dan masyarakat sekitar (Novartis Indonesia, 2014). 2. 1. 3. 4 Sumbangan Korban Bencana Alam PT. Novartis Indonesia berkontribusi terhadap perawatan paliatif, hal ini ditunjukkan dengan memberikan bantuan berupa 3 Ipad kepada Yayasan Rumah Rachel untuk mendukung kegiatan operasional mereka sehari-hari. Perawatan paliatif merupakan komponen penting dalam perawatan kesehatan, khususnya untuk pasien yang mengidap penyakit yang mengancam jiwa. Bantuan berupa perangkat multimedia ini diharapkan dapat menjadi sarana bagi pasien, keluarga pasien, relawan, masyarakat, serta praktisi kesehatan publik untuk mengakses informasi kesehatan, dalam membantu perawatan pada pasien. Selain itu, bantuan ipad ini diharapkan dapat berfungsi sebagai alat bagi pasien untuk penyimpanan catatan, informasi kesehatan seperti obat-obatan, teknologi terbaru, akademis dan publikasi (Novartis Indonesia, 2014). Dengan pemberian donasi ini, Novartis berharap Yayasan Rumah Rachel lebih meningkatkan kesadaran masyarakat akan pentingnya perawatan paliatif terhadap pasien, dan terus menyediakan perawatan ini untuk membantu meningkatkan kualitas hidup pasien di indonesia (Novartis Indonesia, 2014). 2. 2 Struktur Organisasi PT. Novartis Indonesia PT. Novartis Indonesia berlokasi di dua tempat, yaitu Kantor Pusat dan Pemasaran Jakarta yang beralamat di AXA Tower lantai 25-26 Jl. Prof. Dr. Satrio Kav. 18 Kuningan City, Setiabudi Jakarta Selatan 12940. Sedangkan pabrik PT. Novartis Indonesia beralamat di Jl. TB. Simatupang Kp. Gedong, Pasar Rebo Jakarta Timur 13760. Kantor pusat atau Head Office berfokus pada pemasaran produk serta administrasi produk dan perusahaan, sedangkan pabrik berfokus pada proses pembuatan produk mulai dari bahan mentah hingga terbentuk produk jadi Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 8 yang telah dikemas. Oleh karena itu, antara head office dan pabrik memiliki struktur organisasi yang berbeda. 2. 2. 1 Struktur Organisasi Head Office PT. Novartis Indonesia CEO BU (Neuroscience and ophtamology) BU (Integrated Hospital care and critical care) chief of scientific officer chief of finance officer BU (Primary care) BU (Pain and Bone) MSO & BDSL Legal and compliance Market access and Goverment affairs Gambar 2.1. Struktrur Organisasi Head Office PT. Novartis Indonesia 2. 2. 2 Struktur Organisasi Medical and Regulatory Department Medical dan regulatory department merupakan departemen yang ada di head office PT. Novartis Indonesia yang berkonsentrasi pada produk obat secara scientific, termasuk proses regulasi produk. Struktur organisasi dari Medical dan regulatory department dapat dilihat pada Lampiran 1. 2. 2. 2. 1 DRA (Drug Regulatory Affair) DRA merupakan salah satu divisi di bawah Medical and Regulatory department. Divisi ini memiliki fungsi sebagai penghubung antara pihak perusahaan dengan regulatori pemerintah, dalam hal ini adalah BPOM (Badan Pengawas Obat dan Makanan). Tugas utama dari divisi ini adalah untuk melakukan pendaftaran terhadap obat yang akan dipasarkan di Indonesia dan pendaftaran terhadap perubahan mengenai obat-obat yang telah terdaftar dan dipasarkan di Indonesia. Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 9 Secara garis besar tugas DRA di Novartis Indonesia dapat dibagi menjadi dua. Tugas yang pertama adalah registrasi obat baru. Registrasi obat baru dilakukan untuk obat-obatan yang belum mendapatkan nomor izin edar. Staf yang melakukan registrasi obat baru ini harus mengerti dan memahami mengenai obat yang akan didaftarkan, baik dari segi mutu maupun efektivitas dan keamanan obat tersebut. Tugas yang kedua adalah registrasi variasi. Staf yang melakukan registrasi variasi ini dibagi menjadi 2 bagian, yaitu staf registrasi variasi terkait penandaan (safety update) dan variasi terkait mutu (quality update). Staf yang melakukan registrasi variasi harus mampu menentukan apakah pada suatu perubahan produk yang diajukan oleh pihak internal perlu dilakukan registrasi atau tidak, jika perlu maka harus dapat ditentukan termasuk kedalam kategori registrasi apa perubahan yang diajukan tersebut. Di luar tugas utama dalam melakukan registrasi, DRA juga merupakan bertugas sebagai jembatan antara BPOM dengan perusahaan. Misalnya, jika terdapat perubahan yang telah disetujui oleh BPOM, DRA harus segera menginformasikannya dengan pihak perusahaan. Begitu pula pada saat sebelum registrasi, DRA harus menyiapkan dokumen produk obat yang dibutuhkan sesuai dengan persyaratan BPOM. 2. 2. 2. 2 CRA (Clinical Research Association) CRA merupakan divisi yang bertanggung jawab terhadap uji klinik yang dilakukan oleh PT. Novartis Indonesia. Tugas utama dari CRA yaitu memonitor dan memastikan bahwa uji klinik dilaksanakan dengan baik. Hal utama yang menjadi perhatian dari CRA dalam pelaksanaan uji klinik adalah keamanan pasien dan juga data yang diperoleh valid dan akurat. CRA terdiri dari beberapa bagian, yaitu ICRO (International Clinical Research Organization), merupakan manajer atau kepala dari bagian CRA, staf interventional study dan staf observational study. Staf interventional study memiliki tanggung jawab memonitor jalannya uji klinik, mulai dari dilakukannya feasibility study hingga uji klinik selesai dilakukan (site closed out). Staf observational study memiliki tugas utama melakukan studi klinik fase empat dengan mengawasi obat-obatan yang sudah Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 10 disetujui oleh BPOM. Bagian ini akan mengawasi peredaran obat-obatan yang sudah ada di pasaran dan telah digunakan oleh masyarakat. Bentuk pengawasannya dapat berupa melakukan post-marketing surveillance atau melakukan suatu outcome study untuk meneliti bagaimana efek penggunaan obat yang ada di pasaran dengan cara melakukan mega trial. Mega trial umumnya dilakukan pada pemantauan obat-obat pada penyakit yang prevalensinya tinggi di Indonesia, misalnya obat hipertensi atau diabetes. Mega trial menggunakan jumlah subjek yang sangat besar. Pada uji ini, diteliti apakah suatu obat yang telah beredar di pasaran mampu menurunkan angka morbiditas dan mortalitas dari penyakit. Misanya, obat A untuk penyakit diabetes apakah setelah dikonsumsi oleh pasein dalam jangka waktu tertentu mampu menurunkan angka kejadian gagal ginjal. Selain kedua hal tersebut, pengawasan juga dapat dilakukan dengan mengumpulkan data pemakaian obat dari dokter. 2. 2. 2. 3 MA (Medical Affair) Medical affair (MA) merupakan divisi yang bertanggung jawab dalam membangun hubungan peer to peer dengan KOL (Key Opinion Leader), memfasilitasi interaksi antara dokter dan representatif dari perusahaan dan menyediakan pemahaman dari KOL mengenai obat dan penyakit di masyarakat. KOL merupakan dokter-dokter yang kompeten di bidangnya masing-masing yang pendapatnya menjadi rujukan bagi dokter lainnya. Dalam melaksanakan tugasnya ini, MA dari PT. Novartis Indonesia dibagi ke dalam 4 bagian berdarkan ruang lingkup produk obat yang ditangani, yaitu Established medicine, NSO (Neuroscience and Ophtalmology), Primary care dan IHC&CC (Integrated Hospital Care and Critical Care). Pada setiap bagian terdiri dari Medical Advisor dan Medical Scientific Liaison (MSL). Medical Advisor memiliki fungsi strategik dalam MA. Tugas dari Medical Advisor yaitu: a. Melakukan pendekatan ke KOL Medical advisor memiliki peran dalam merencanakan input dari para KOL yang harus didapatkan oleh MSL ketika melakukan pendekatan. Selain itu, juga memastikan mengenai materi yang harus diterima oleh para KOL sesuai Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 11 dengan yang diharapkan. Medical Advisor ini yang memonitor kerja dari MSL. b. Pelatihan Pelatihan yang diadakan dapat berupa training internal atau training eksternal. Training eksternal diadakan untuk para KOL. c. PMS (Post-Marketting Surveilance) d. Menjawab medical inqueries e. Review produk Tugas utama dari Medical Scientific Liaison (MSL) adalah membangun hubungan dengan para KOL. MSL adalah staf yang langsung berhubungan dengan KOL untuk melakukan pendekatan dan mendapatkan opini dan insight dari para KOL mengenai produk-produk yang akan maupun telah dipasarkan. Selain itu, para MSL juga ikut membantu CRA dalam melakukan proses pengawasan paska pemasaran terhadap suatu produk. Setiap MSL akan menangani satu produk. Produk-produk yang memiliki MSL ini adalah produkproduk yang sebelumnya telah dikaji bahwa obat ini membutuhkan pendapat scientific dari para ahli. 2. 2. 2. 4 PV (Pharmacovigilans) & RMP (Risk Management Plan) PV & RMP merupakan satu bagian di Medical and Regulatory Department. Bagian ini bertanggung jawab terhadap pemantauan pemakaian suatu produk obat. Pemantauan terhadap penggunaan obat dimulai dari uji pra-klinik hingga uji klinik fase 4 dilakukan. Pemantauan ini dilakukan untuk membantu pelaksaan RMP, yaitu perencanaan untuk meminimalkan resiko dalam penggunaan obat. Pelaporan terhadap pemakaian suatu produk obat dapat berupa pelaporan terhadap timbulnya efek obat yang tidak diinginkan maupun keluhan lain dari produk yang berhubungan dengan keamanan produk. Terdapat beberapa sumber pelaporan, yaitu secara spontan, laporan dari hasil uji klinik, program marketing, maupun berdasarkan kasus yang ditemukan dari literatur. Terdapat empat kriteria suatu laporan dinilai lengkap, yaitu adanya pelapor, produk yang digunakan, keluhan yang dirasakan, dan dilakukannya Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 12 pelaporan. Setiap laporan yang diterima, sebelumnya harus ada inform consent terhadap hal yang dilaporkannya, dan harus dilakukan secara sukarela oleh pelapornya.. Dari laporan yang diterima, kemudian dibuat laporan tertulis kepada BPOM dengan rentang waktu maksimal 5 hari setelah laporan tersebut diterima oleh divisi ini. 2. 2. 2. 5 QA (Quality Assurance) Head Office QA yang berada di Head office (HO) memiliki tugas yang berbeda dari QA yang berada di pabrik. Bagian Quality Assurance yang berada di HO memiliki tugas untuk melakukan pengawasan terhadap kualitas produk setelah dipasarkan. Quality Assurance HO akan mengumpulkan dan menangani berbagai pelaporan mengenai produk yang telah dipasarkan dari segi mutu. Untuk pembagian tugas kerjanya, QA ini dibagi menjadi 2, yaitu QA development dan QA GMP. QA development akan berhubungan dengan pihak-pihak clinical research association, regulatory affairs, dan medical affairs. Sedangkan QA GMP mempunyai fungsi untuk melakukan monitoring ke pabrik. Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 13 BAB 3 TINJAUAN KHUSUS 3. 1 Kriteria dan Tata Laksana Registrasi Obat 3. 1. 1 Registrasi Registrasi adalah prosedur pendaftaran dan evaluasi suatu produk untuk mendapatkan izin edar. Izin edar adalah suatu bentuk persetujuan registrasi untuk dapat diedarkan di wilayah Indonesia. Registrasi bertujuan untuk melindungi masyarakat dari peredaran produk yang tidak memenuhi persyaratan khasiat, keamanan, dan mutu. Registrasi terdiri atas registrasi baru, registrasi variasi, dan registrasi ulang. Registrasi baru adalah registrasi produk yang belum mendapatkan izin edar di Indonesia. Registrasi variasi adalah registrasi perubahan aspek apapun pada produk yang telah memiliki izin edar di Indonesia, termasuk tetapi tidak terbatas pada perubahan formulasi, metoda, proses pembuatan, spesifikasi untuk obat dan bahan baku, wadah, kemasan dan penandaan (Badan POM RI, 2011). Registrasi Variasi Major (VaMa) adalah registrasi variasi yang berpengaruh bermakna terhadap aspek khasiat, keamanan, dan/atau mutu obat. Registrasi Variasi Minor yang Memerlukan Persetujuan (VaMi-B) adalah registrasi variasi yang tidak termasuk kategori registrasi variasi minor dengan notifikasi maupun variasi major. Registrasi Variasi Minor dengan Notifikasi (VaMi-A) adalah registrasi variasi yang berpengaruh minimal atau tidak berpengaruh sama sekali terhadap aspek khasiat, keamanan, dan/atau mutu obat, serta tidak merubah informasi pada sertifikat izin edar. Registrasi ulang adalah registrasi perpanjangan masa berlaku izin edar (Badan POM RI, 2011). 3. 1. 2 Pendaftar Pendaftar adalah industri farmasi yang telah mendapat izin industri farmasi sesuai ketentuan perundang-undangan (Badan POM RI, 2011). Setiap pendaftar bertanggung jawab atas kelengkapan dokumen yang diserahkan, kebenaran semua informasi yang tercantum dalam dokumen registrasi, kebenaran dan keabsahan dokumen yang dilampirkan untuk kelengkapan registrasi, dan perubahan data dan informasi dari produk yang sedang dalam proses registrasi 13 Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia 14 atau sudah memiliki izin edar. Jenis pendaftar dibagi menjadi beberapa kategori sesuai produk yang didaftarkan, yaitu (Badan POM RI, 2011) : a. Pendaftar Produk yang Diproduksi di Dalam Negeri Produk dalam negeri, meliputi produk tanpa lisensi, produk lisensi, dan produk kontrak. Pendaftar produk tanpa lisensi adalah pendaftar yang memiliki izin industri farmasi dan memiliki sertifikat Cara Pembuatan Obat yang Baik (CPOB) yang masih berlaku sesuai dengan jenis dan bentuk sediaan yang diregistrasikan. Pendaftar produk lisensi adalah penerima lisensi yang memiliki ketentuan seperti pendaftar tanpa lisensi dan dokumen perjanjian lisensi. Pendaftar obat kontrak adalah pemberi kontrak yang memiliki izin industri farmasi, paling sedikit satu fasilitas produksi sediaan lain yang telah memenuhi persyaratan CPOB, dan dokumen perjanjian kontrak. b. Pendaftar Produk impor Pendaftar produk impor adalah industri farmasi dalam negeri yang mendapat persetujuan tertulis dari industri farmasi di luar negeri. Industri pemilik produk di luar negeri wajib memiliki izin industri farmasi dan memenuhi persyaratan CPOB yang dibuktikan dengan sertifikat CPOB yang masih berlaku atau dokumen lain yang setara, dan data inspeksi terakhir atau perubahan terkait paling lama dua tahun yang dikeluarkan oleh otoritas pengawas obat setempat dan/atau otoritas pengawas obat negara lain. Pendaftar juga harus menyerahkan Dokumen Induk Farmasi atau SMF (Site Master File) terbaru jika industri farmasi di luar negeri belum mempunyai produk dengan jenis dan bentuk sediaan yang sama dengan yang disetujui beredar di Indonesia atau industri tersebut mempunyai produk yang beredar di Indonesia dengan jenis dan bentuk sediaan yang sama namun terjadi perubahan pada fasilitas produksi. c. Pendaftar Produk Khusus Ekspor Pendaftar produk khusus ekspor adalah industri farmasi terdiri dari pendaftar produk dalam negeri yang ditujukan khusus ekspor dan produk impor khusus ekspor. Produk khusus ekspor dilarang diedarkan di wilayah Indonesia. d. Pendaftar Produk yang Dilindungi Paten Pendaftar produk yang dilindungi paten adalah pemilik hak paten atau yang ditunjuk oleh pemilik hak paten. Pendaftaran produk yang masih dilindungi paten Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 15 dapat dilakukan oleh pendaftar yang bukan pemilik hak paten sesuai dengan ketentuan perundang-undangan. Pendaftaran dapat diajukan mulai dua tahun sebelum berakhirnya perlindungan paten dengan melampirkan informasi tanggal berakhirnya perlindungan paten dan data ekivalensi untuk menjamin kesetaraan khasiat, keamanan, dan mutu. 3. 1. 3 Registrasi Obat 3. 1. 3. 1 Kategori Registrasi Obat Registrasi obat terdiri atas registrasi baru, registrasi variasi, dan registrasi ulang. Registrasi baru diawali dengan proses pra-registrasi. Registrasi baru terdiri atas tiga kategori, yaitu (Badan POM RI, 2011): a. Kategori 1 untuk registrasi obat baru dan produk biologi, temasuk produk biologi sejenis (PBS)/Similar Biotherapic Product (SBP), meliputi: 1. Registrasi obat baru dengan zat aktif baru atau produk biologi 2. Registrasi obat baru atau produk biologi dengan kombinasi baru 3. Registrasi obat baru atau produk biologi dengan bentuk sediaan baru atau kekuatan baru 4. Registrasi obat baru atau produk biologi dengan rute pemberian baru 5. Registrasi produk biologi sejenis (PBS)/Similar Biotherapic Product (SBP) b. Kategori 2 untuk registrasi obat copy, meliputi: 1. Registrasi obat copy yang memerlukan uji klinik 2. Registrasi obat copy yang tidak memerlukan uji klinik c. Kategori 3 : registrasi sediaan lain yang mengandung obat Registrasi variasi dilakukan apabila terjadi perubahan terhadap obat yang telah mendapat NIE. Registrasi variasi terdiri atas tiga kategori, yaitu : a. Kategori 4 : registrasi variasi major (VaMa) b. Kategori 5 : registrasi variasi minor yang memerlukan persetujuan (VaMi-B) c. Kategori 6 : registrasi variasi minor dengan notifikasi (VaMi-A) Permohonan pengajuan registrasi ulang dilakukan paling cepat 120 hari sebelum berakhir masa berlaku izin edar. Permohonan ini diajukan dengan mengisi formulir registrasi (Lampiran 2) dan melampirkan dokumen registrasi ulang. Persetujuan atas permohonan registrasi ulang secara otomatis berlaku sejak Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 16 berakhir masa izin edarnya, kecuali untuk registrasi ulang dengan informasi terbaru yang terkait aspek keamanan obat, khasiat obat, dan/atau kerasionalan formula obat. Registrasi ulang termasuk dalam kategori 7. 3. 1. 3. 2 Tata Laksana Registrasi Obat Permohonan pra registrasi dan registrasi diajukan oleh pendaftar secara tertulis kepada Kepala BPOM dan dilampiri dengan dokumen pra registrasi atau dokumen registrasi. Isi dokumen proses pra registrasi atau registrasi dapat dilihat pada Lampiran 3. Proses registrasi dibagi ke dalam dua tahap, yaitu tahap pra registrasi dan tahap registrasi (Badan POM RI, 2011). a. Tahap Pra Registrasi Permohonan pra registrasi dilakukan untuk penapisan registrasi obat, penentuan kategori registrasi, penentuan jalur evaluasi, penentuan biaya evaluasi, dan penentuan dokumen registrasi obat. Permohonan ini diajukan dengan mengisi formulir pra registrasi, menyerahkan bukti pembayaran biaya pra registrasi, dan melampirkan dokumen lengkap pra registrasi. Alur pelaksanaan pra-registrasi dapat dilihat pada Lampiran 4. Pada tahap ini, paling lama dalam jangka waktu 40 hari sejak diterimanya permohonan pra registrasi Kepala BPOM memberikan surat hasil pra registrasi (HPR) kepada pendaftar yang berlaku satu tahun sejak tanggal dikeluarkan. Apabila sebelum jangka waktu yang dimaksud diperlukan penambahan data atas dokumen administratif dan/atau teknis, maka pendaftar akan diberikan surat permintaan tambahan data. Perhitungan jangka waktu pengeluaran HPR diberhentikan (clock off) sampai pendaftar menyampaikan tambahan data yang diterima dan penyerahan tambahan data tersebut harus disampaikan paling lama 20 hari setelah surat dikeluarkan. Jalur evaluasi untuk tahap pra registrasi terdiri atas jalur 40 hari, meliputi registrasi variasi minor yang memerlukan persetujuan dan registrasi obat khusus ekspor, jalur 100 hari meliputi (Badan POM RI, 2011) : 1) Registrasi baru obat baru dan produk biologi yang diindikasikan untuk terapi penyakit serius yang mengancam nyawa manusia (life saving), dan/atau mudah menular pada orang lain, dan/atau belum ada atau kurangnya pilihan terapi lain yang aman dan efektif. Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 17 2) Registrasi baru obat baru dan produk biologi yang berdasarkan justifikasi diindikasikan untuk penyakit serius dan langka (orphan drug). 3) Registrasi baru obat baru dan produk biologi ditujukan untuk program kesehatan masyarakat. 4) Registrasi baru obat baru dan produk biologi yang telah melalui proses obat pengembangan baru yang dikembangkan oleh industri farmasi atau institusi riset di Indonesia dan seluruh tahapan uji kliniknya dilakukan di Indonesia. 5) Registrasi baru obat copy esensial generik yang dilengkapi dengan dokumen penunjang kebutuhan program atau data pendukung sebagai obat esensial. 6) Registrasi baru obat copy dengan standar informasi elektronik (Stinel). 7) Registrasi variasi major indikasi baru/posologi baru untuk obat yang ditujukan sebagaimana dimaksud pada huruf a-d. 8) Registrasi variasi major yang tidak termasuk pada huruf g. Jalur 150 hari meliputi (Badan POM RI, 2011) : 1) Registrasi baru obat baru, produk biologi, dan registrasi variasi major indikasi baru/posologi baru yang telah disetujui di negara yang telah menerapkan sistem evaluasi terharmonisasi dan di negara dengan sistem evaluasi yang telah dikenal baik. 2) Registrasi baru obat baru, produk biologi, dan registrasi variasi major indikasi baru/posologi baru yang telah disetujui paling sedikit di tiga negara dengan sistem evaluasi yang telah dikenal baik. 3) Registrasi baru obat copy tanpa Stinel. Jalur 300 hari, meliputi registrasi baru obat baru, produk biologi, produk biologi sejenis, atau registrasi variasi major indikasi baru/posologi baru yang tidak termasuk dalam jalur evaluasi sebagaimana dimaksud pada jalur 100 dan 150 hari (Badan POM RI, 2011). b. Tahap Registrasi Pengajuan registrasi dilakukan dengan menyerahkan berkas registrasi dengan mengisi formulir registrasi dan disket disertai bukti pembayaran biaya evaluasi dan pendaftaran, serta Hasil Pra Registrasi. Berkas registrasi terdiri Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 18 atas formulir registrasi dengan dokumen administratif dan dokumen penunjang. Dokumen tersebut disusun sesuai format ASEAN Common Technical Dossier (ACTD). Dokumen registrasi yang diserahkan harus dilengkapi dengan rancangan kemasan dan brosur. Rancangan kemasan, meliputi etiket, dus/bungkus luar, strip/blister, catch over, ampul atau vial, dan kemasan lain sesuai ketentuan tentang pembungkusan luar dan penandaan yang berlaku, yang merupakan rancangan kemasan obat yang akan diedarkan, dan dilengkapi dengan rancangan warna (Badan POM RI, 2011). Alur Proses registrasi dapat dilihat pada Lampiran 5. 3. 1. 3. 3 Evaluasi dan Pemberian Keputusan Evaluasi dilakukan terhadap dokumen registrasi yang telah dinyatakan lengkap. Evaluasi dilaksanakan sesuai jalur evaluasi 40 hari kerja, 100 hari kerja, 150 hari kerja, atau 300 hari kerja yang dihitung sejak penyerahan dokumen registrasi obat. Untuk melakukan evaluasi, dibentuk Komite Nasional (KOMNAS) Penilai Obat, Panitia Penilai Khasiat Keamanan, Panitia Penilai Mutu, dan Panitia Penilai Informasi Produk dan Penandaan. Evaluasi data khasiat dan keamanan dilakukan berdasarkan pembuktian ilmiah dan pedoman penilaian khasiat dan keamanan oleh Penilai Khasiat Keamanan. Hasil evaluasi khasiat dan keamanan disampaikan kepada pendaftar paling lambat 30 hari. Berdasarkan hasil evaluasi tersebut, KOMNAS Penilai Obat dapat memberikan rekomendasi kepada Kepala BPOM. Apabila diperlukan klarifikasi dan/atau penjelasan teknis secara rinci dari dokumen yang diserahkan, KOMNAS Penilai Obat dapat merekomendasikan untuk dilakukan dengar pendapat oleh pendaftar. Untuk dengar pendapat, BPOM akan menyampaikan surat pemberitahuan kepada pendaftar. Evaluasi informasi produk dan penandaan dilakukan oleh Penilai Informasi Produk dan Penandaan sesuai kriteria yang lengkap, objektif, tidak menyesatkan yang menjamin penggunaan obat secara tepat, rasional, dan aman. Jika diperlukan tambahan data, maka permintaan tambahan data akan disampaikan kepada pendaftar secara tertulis. Tambahan data ini harus disampaikan paling lama 100 hari kerja setelah tanggal permintaan, sementara itu waktu perhitungan waktu evaluasi dihentikan (clock off). Perhitungan waktu Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 19 evaluasi dilanjutkan setelah pendaftar menyerahkan tambahan data (Badan POM RI, 2011). Keputusan terhadap registrasi obat dapat berupa pemberian persetujuan atau penolakan yang dipertimbangkan berdasarkan hasil evaluasi dokumen registrasi dan hasil pemeriksaan pada pabrik pembuatan obat. Persetujuan diberikan secara tertulis kepada pendaftar berupa persetujuan izin edar, persetujuan impor dalam bentuk ruahan, persetujuan impor khusus ekspor, dan persetujuan khusus ekspor. Penolakan registrasi disampaikan secara tertulis oleh Kepala BPOM berupa surat penolakan dan biaya registrasi yang telah dibayarkan tidak dapat ditarik kembali. Registrasi yang ditolak dapat diajukan kembali dengan mengikuti tata cara sesuai ketentuan. Jika terdapat keberatan terhadap hasil evaluasi khasiat dan keamanan dari KOMNAS Penilai Obat maka pendaftar dapat mengajukan permohonan dengar pendapat secara tertulis dalam jangka waktu 20 hari sejak tanggal surat pemberitahuan kepada Kepala BPOM. Jika keputusan hasil registrasi berupa penolakan, maka pendaftar dapat mengajukan permohonan peninjauan kembali kepada Kepala BPOM. Peninjauan kembali ini dapat diajukan paling lama enam bulan setelah tanggal surat penolakan dan hanya dapat dilakukan satu kali. Permohonan ini harus dilengkapi dengan data baru dan/atau data yang sudah pernah diajukan dengan dilengkapi justifikasi. Pembahasan terhadap surat permohonan ini dilakukan paling lama 100 hari sejak dokumen diterima. Apabila registrasi ditolak, pendaftar dapat mengajukan permohonan registrasi kembali sesuai ketentuan. Akan tetapi jika registrasi ditolak karena alasan tidak memenuhi kriteria khasiat dan keamanan, selain harus mengikuti tata cara sesuai ketentuan, registrasi kembali hanya dapat diajukan dengan data baru dan paling cepat satu tahun setelah tanggal surat penolakan (Badan POM RI, 2011). 3. 1. 3. 4 Masa Berlaku dan Pelaksanaan Izin Edar Izin edar obat berlaku paling lama lima tahun selama masih memenuhi ketentuan yang berlaku termasuk persetujuan impor dalam bentuk ruahan, persetujuan impor khusus ekspor, dan persetujuan khusus ekspor. Jika obat yang diregistrasikan berdasarkan perjanjian atau penunjukkan dengan masa kerja sama kurang dari lima tahun, maka masa berlaku izin edar disesuaikan dengan masa Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 20 berlaku kerja sama dalam dokumen perjanjian. Dalam hal perjanjian atau penunjukkan kerja sama dihentikan sebelum masa izin edar berakhir, izin edar obat yang bersangkutan dibatalkan. Obat yang telah habis masa berlaku izin edarnya dapat diperpanjang selama memenuhi kriteria melalui mekanisme registrasi ulang. Apabila obat yang telah habis masa berlaku izin edarnya dan tidak diperpanjang maka dianggap sebagai obat yang tidak memiliki izin edar (Badan POM RI, 2011). Pendaftar wajib memproduksi atau mengimpor, dan mengedarkan obat yang telah mendapatkan izin edar selambatnya satu tahun setelah tanggal persetujuan dikeluarkan dan harus melapor kepada Kepala BPOM dengan menyerahkan kemasan siap edar. Kemasan siap edar yang diserahkan berupa kemasan primer, kemasan sekunder, dan informasi produk. Penyerahan kemasan dilakukan paling lambat satu bulan sebelum pelaksanaan peredaran obat. Pemilik izin edar obat wajib melakukan pemantauan khasiat, keamanan, dan mutu selama obat diedarkan dan melaporkan hasilnya kepada Kepala BPOM (Badan POM RI, 2011). 3. 1. 3. 5 Evaluasi Kembali dan Sanksi Evaluasi kembali dapat dilakukan terhadap obat yang telah mendapat izin edar. Evaluasi ini dilakukan jika berdasarkan hasil pemantauan terdapat perkembangan baru mengenai khasiat, keamanan, dan mutu obat yang berbeda dari data penunjang saat registrasi. Keputusan hasil evaluasi kembali dapat berupa perubahan penandaan, perbaikan komposisi/ formula, pemberian batasan penggunaan, penarikan obat dari peredaran, dan/ atau pembekuan izin edar dan/atau pembatalan izin edar (Badan POM RI, 2011). Pendaftar yang tidak memenuhi ketentuan dapat dikenakan sanksi administratif berupa peringatan tertulis, pembatalan proses registrasi obat, pembekuan izin edar obat yang bersangkutan, pembatalan izin edar obat yang bersangkutan, atau sanksi administratif lain sesuai ketentuan perundang undangan. Pemberian sanksi berupa pembatalan atau pembekuan izin edar terjadi jika tidak melaksanakan kewajiban memproduksi, mengimpor, atau mengedarkan obat yang telah mendapat izin edar, selama 12 bulan berturut-turut tidak memproduksi, mengimpor, atau mengedarkan, izin industri farmasi pemilik izin edar dicabut, Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 21 dan/ atau pemilik izin edar melakukan pelanggaran di bidang produksi dan/ atau distribusi obat. Pembekuan dan pembatalan izin edar dilakukan secara tertulis kepada pemilik izin edar (Badan POM RI, 2011). 3. 2 ASEAN Common Technical Dossier / ASEAN Common Technical Requirements ASEAN Common Technical Dossier (ACTD) adalah format umum yang digunakan untuk menyusun dokumen registrasi yang akan didaftarkan kepada badan regulasi obat di wilayah ASEAN. Tujuan penggunaan ACTD adalah agar penggambaran berbagai informasi produk menjadi transparan dan tidak ambigu, sehingga mempermudah pemeriksaan data-data dasar dan membantu pembaca menjadi lebih cepat terorientasi kepada isi pendaftaran produk (Badan POM RI, 2011). ASEAN Common Technical Requirements (ACTR) adalah seluruh materi tertulis yang bertujuan untuk membantu pendaftar untuk menyiapkan dokumen registrasi secara konsisten sesuai dengan harapan seluruh badan otoritas regulasi obat di ASEAN. ACTR ini berisi seluruh persyaratan dan parameter-parameter yang harus dipenuhi oleh produk obat, baik dari segi kualitas, keamanan, dan efikasi. Untuk memenuhi persyaratan yang tercantum dalam ACTR, maka dibuat berbagai pedoman, seperti pedoman uji stabilitas, validasi analisis, validasi proses, validasi uji bioekivalensi, dan berbagai pedoman keamanan dan pedoman efikasi (Badan POM RI, 2011). 3. 2. 1 Ketentuan ACTD Teks dan tabel harus disiapkan dengan margin yang memungkinkan dokumen dapat dicetak dengan baik pada kertas berukuran A4. Margin kiri sebaiknya cukup besar sehingga informasi tidak bias dengan menggunakan metode binding. Jenis huruf dan besar huruf adalah Times New Roman 12. Untuk teks dan tabel, sebaiknya jenis dan ukuran huruf yang cukup besar sehingga mudah dibaca bahkan setelah difotokopi. Setiap halaman harus diberi angka dengan halaman pertama pada setiap bagian disebut sebagai halaman 1. Untuk singkatan dan pengertian Common Technical Dossier harus dijelaskan setiap pertama kali digunakan pada setiap bagian. Referensi harus dicantumkan menurut Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 22 1979 Vancouver Declaration of Uniform requirements for Manuscript Submitted to Biomedical Journals (Badan POM RI, 2011). 3. 2. 2 Pembagian ACTD ACTD dibagi ke dalam 4 bagian, yaitu Dokumen Administratif, Dokumen Mutu, Dokumen Non klinik, dan Dokumen Klinik (Badan POM RI, 2011). 3. 2. 2. 1 Dokumen Administratif Dokumen ini berisi pengenalan umum dari sediaan farmasi yang akan didaftarkan. Pada bagian awal dokumen ini berisi keseluruhan tabel isi atau keseluruhan dokumen ACTD untuk memberikan informasi dasar yang dapat dicari langsung. Selanjutnya, dokumen ini berisi data administratif yang memerlukan dokumentasi spesifik sejelas mungkin, yaitu formulir pendaftaran, label, brosur, kemasan, dan lain-lain. Bagian akhir dari dokumen ini adalah informasi produk yang memberikan informasi seperlunya, meliputi informasi pemberian obat, mekanisme kerja, efek samping, dan sebagainya. Isi dari dokumen administratif dapat dilihat pada Lampiran 3 (Badan POM RI, 2011). 3. 2. 2. 2 Dokumen Mutu Bagian ini berisikan penjelasan mengenai kualitas produk obat secara menyeluruh beserta laporan penelitiannya. Dokumen kontrol kualitas harus dijelaskan sejelas mungkin. Isi dari dokumen mutu dapat dilihat pada Lampiran 2 (Badan POM RI, 2011). 3. 2. 2. 3 Dokumen Non klinik Bagian ini harus memberikan penjelasan non klinik yang disertai dengan rangkuman non klinik tertulis dan rangkuman non klinik dalam bentuk tabel. Data dalam bagian ini tidak disertakan pada pendaftaran produk generik, produk yang telah memiliki NIE dengan variasi minor, dan juga pada beberapa produk dengan variasi mayor. Untuk negara-negara anggota ASEAN tertentu, laporan penelitian dari bagian ini mungkin tidak dibutuhkan untuk produk dengan zat kimia baru (New Chemical Entity/NCE), produk bioteknologi, dan produk dengan variasi mayor lain jika produk aslinya telah teregistrasi dan sudah disetujui Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 23 untuk izin pemasaran di negara asalnya. Isi dari dokumen non klinik dapat dilihat pada Lampiran 3 (Badan POM RI, 2011). 3. 2. 2. 4 Dokumen Klinik Bagian ini memberikan penjelasan klinik dan rangkuman klinik. Dokumen dari bagian ini juga tidak disertakan pada pendaftaran produk generik, produk yang telah memiliki NIE dengan variasi minor, dan juga pada beberapa produk dengan variasi mayor. Untuk negara-negara anggota ASEAN, laporan penelitian dari bagian ini mungkin tidak dibutuhkan untuk produk dengan zat kimia baru (NCE), produk bioteknologi, dan produk dengan variasi mayor lain jika produk aslinya telah teregistrasi dan sudah disetujui untuk izin pemasaran di Negara asalnya. Isi dari dokumen klinik dapat dilihat pada Lampiran 3 (Badan POM RI, 2011). Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 24 BAB 4 PEMBAHASAN 4. 1 Registrasi Obat Registrasi adalah proses yang harus dilakukan untuk untuk mendapatkan nomor ijin edar (NIE) suatu obat sehingga obat tersebut dapat diedarkan di wilayah Indonesia. Untuk mendapatkan nomor izin edar suatu obat harus memiliki kriteria, antara lain memiliki khasiat yang meyakinkan dan keamanan memadai dibuktikan melalui uji non-klinik dan uji klinik atau bukti-bukti lain sesuai dengan standar perkembangan ilmu pengetahuan yang bersangkutan, obat memenuhi syarat yang dinilai dari proses produksi sesuai dengan Cara Pembuatan Obat yang Baik (CPOB), spesifikasi dan metode analisis terhadap semua bahan yang digunakan serta produk jadi dengan bukti yang sahih, obat memiliki penandaan dan informasi produk yang berisi informasi lengkap, objektif dan tidak menyesatkan yang dapat menjamin penggunaan obat secara tepat, rasional dan aman, obat sesuai dengan kebutuhan nyata masyarakat, dan khusus untuk obat psikotropika baru harus memiliki keunggulan dibandingkan dengan obat yang telah disetujui beredar di Indonesia, dan untuk kontrasepsi atau obat lain yang digunakan dalam program nasional dapat dipersyaratkan uji klinik di Indonesia (Badan POM RI, 2011). Registrasi bertujuan untuk melindungi masyarakat dari peredaran produk yang tidak memenuhi persyaratan khasiat, keamanan, dan mutu. Pengajuan registrasi obat ditujukan kepada Kepala Badan dan selanjutnya dilakukan evaluasi oleh evaluator yang ditunjuk dan ahli dibidangnya untuk menilai obat tersebut telah sesuai dengan persyaratan yang ada atau tidak. Di Indonesia, evaluasi dokumen registrasi obat dilakukan oleh BPOM selaku pihak regulator. Tata lakasana registrasi obat diatur oleh Badan POM dalam keputusan Kepala Badan Pengawas Obat dan Makanan Republik Indonesia Nomor HK.03.1.23.10.11.08481 Tahun 2011 Tentang Kriteria dan Tata Laksana Registrasi Obat menggantikan Kriteria dan Tata Laksana Registrasi Obat dalam Keputusan Kepala Badan Pengawas Obat dan Makanan Nomor HK.00.05.3.1950 tahun 2003 karena telah tidak sesuai lagi dengan perkembangan ilmu 24 Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia 25 pengetahuan dan teknologi. Perubahan yang terjadi juga merupakan penyempurnaan dan penyesuaian dengan regulasi terbaru di Indonesia dan ASEAN, namun secara garis besar isi dari regulasi mengenai tata cara registrasi obat tidak terlalu banyak berubah. Perubahan yang terdapat pada terjadi pada tata laksana registrasi obat tahun 2011 dari tahun 2003 antara lain adalah penjelasan mengenai pendaftar registrasi yang sebelumnya diatur bahwa pendaftar merupakan industri farmasi yang memiliki izin industri dan PBF namun sekarang hanya industi farmasi yang telah memiliki izin industri; terdapat perubahan kategori registrasi yang sebelumnya ada 10 kategori namun sekarang hanya 7 kategori registrasi saja; terdapat perubahan pada jalur pra-registrasi yang sebelumnya hanya ditujukan untuk obat yang belum memiliki izin edar namun sekarang pra-registrasi dilakukan pada obat baru dan obat copy yang belum pernah didaftarkan dan pada registrasi variasi yang termasuk kategori variasi mayor; terdapat perubahan pengelompokan registrasi variasi yang sebelumnya tidak ada pengelompokan registrasi variasi namun sekarang menjadi tiga kelompok registrasi yaitu registrasi variasi mayor, registrasi variasi minor yang memerlukan persetujuan dan registrasi variasi minor dengan notifikasi; terdapat ketentuan bolar provision yakni obat paten yang sebelumnya tidak boleh diregistrasikan namun sekarang ada ketentuan yang menyebutkan bahwa obat paten diizinkan untuk dilakukan registrasi; dan terdapat perubahan jalur evaluasi. 4. 2 Registrasi Obat Baru Obat baru adalah obat yang mengandung zat aktif baru, zat tambahan baru, bentuk sediaan atau rute pemberian baru, kekuatan baru, atau kombinasi baru yang belum pernah disetujui di Indonesia (Badan POM RI, 2011). Registrasi obat baru termasuk dalam golongan registrasi baru kategori 1. Proses registrasi obat baru terbagi ke dalam dua tahap, yaitu tahap pra registrasi dan kemudian dilanjutkan dengan tahap registrasi. Tahapan pra registrasi dilakukan untuk menentukan kategori registrasi, jalur evaluasi, dan biaya evaluasi pada tahapan registrasi selanjutnya. Kegiatan pra registrasi diawali dengan pengajuan pendaftaran secara online melalui https://www.antrianobat.co untuk mendapatkan Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 26 jadwal layanan di minggu berikutnya. Jadwal pendaftaran dan nomor antrian akan dikirimkan melalui email setiap jumat. Pendaftar kemudian melakukan verifikasi kelengkapan dokumen sesuai jadwal yang telah ditetapkan. Kelengkapan dokumen dan persyaratan untuk registrasi baru dapat dilihat pada Lampiran 6. Permohonan pra registrasi dan registrasi diajukan oleh pendaftar secara tertulis kepada Kepala BPOM dan dilampiri dengan dokumen sesuai tahapannya. Secara garis besar dokumen registrasi yang harus diserahkan terdiri dari 4 bagian, yaitu: a. Dokumen administratif b. Dokumen mutu c. Dokumen non-klinik d. Dokumen klinik Kelengkapan dokumen tersebut disatukan dalam ordner/map berwarna biru. Selain kelengkapan dokumen dan persyaratan seperti yang tertera pada Lampiran 6, untuk obat kategori 1 atau obat baru, pendaftar juga harus menyerahkan rencana manajemen risiko yang kemudian akan ditetapkan. Dokumen pra-registrasi atau registrasi yang telah disiapkan akan di verifikasi pada bagian loket pra registrasi. Setelah dinyatakan lengkap oleh petugas desk evaluator dengan bukti cap lengkap pada dokumen checklist selanjutnya pendaftar akan mendapat surat perintah bayar untuk biaya evaluasi pra registrasi. Setelah pembayaran dilakukan, dokumen-dokumen tersebut beserta bukti cap lengkap, bukti bayar dan formulir pra registrasi yang sebelumnya telah diisi di masukkan ke dalam loket bagian pemasukan dokumen untuk dilakukan validasi kelengkapan dokumen. Bersamaan dengan dimasukkannya dokumen pra registrasi pendaftar akan mendapatkan surat tanda terima dokumen. Setelah proses tersebut, pendaftar harus terus memantau perkembangan tahapan pra registrasi yang sedang berjalan dan harus siap jika pada tahapan tersebut diperlukan data tambahan. Hasil pra registrasi akan keluar dalam jangka waktu 40 hari kerja terhitung sejak dokumen dimasukkan. Setelah 40 hari kerja, pendaftar akan mendapatkan hasil pra registrasi, di dalamnya akan mencantumkan kategori registrasi, jalur evaluasi, biaya evaluasi tahapan Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 27 registrasi serta pengembalian dokumen pra registrasi atau registrasi. Tahapan selanjutnya yaitu registrasi. Pada tahapan ini hampir sama dengan tahapan pra registrasi. Perbedaan terdapat pada saat dilakukannya validasi kelengkapan dokumen. Saat melakukan validasi kelengkapan dokumen registrasi pendaftar harus menyertakan hasil pra registrasi, bukti bayar, cap bukti lengkap, disc beserta dokumen registrasi serta mengisi formulir permohonan registrasi dan formulir pengisian disc. Setelah pembayaran dilakukan dokumen-dokumen tersebut beserta bukti cap lengkap, bukti bayar, formulir registrasi, hasil pra registrasi, dan disc yang sebelumnya telah diisi di masukkan ke dalam loket bagian pemasukan dokumen. Bersamaan dengan dimasukkannya dokumen registrasi pendaftar akan mendapatkan surat tanda terima dokumen. Setelah proses tersebut, pendaftar harus terus memantau perkembangan tahapan registrasi yang sedang berjalan dan harus siap jika pada tahapan tersebut diperlukan data tambahan. Hasil registrasi akan keluar dalam jangka waktu sesuai dengan kategori registrasi, terhitung sejak dokumen dimasukkan. Setelahnya, jika telah disetujui, maka akan didapatkan nomor izin edar (NIE) yang berlaku selama 5 tahun, dan dapat diperpanjang jika telah habis masa berlakunya dengan registrasi ulang. 4. 3 Registrasi Obat Copy Obat copy adalah obat yang mengandung zat aktif dengan komposisi, kekuatan, bentuk sediaan, rute pemberian, indikasi dan posologi sama dengan obat yang sudah disetujui. Untuk kelengkapan dokumen registrasi obat copy secara umum memiliki kesamaan dengan registrasi obat baru, namun terdapat perbedaan pada beberapa bagian, diantaranya yaitu: untuk registrasi obat copy tidak disertakan bagian 3, yaitu dokumen non-klinik, serta perbedaan warna ordner/map yang digunakan. Untuk pra registrasi obat copy menggunakan ordner/map hitam dan untuk registrasinya menggunakan ordner/map merah. 4. 4 Registrasi Variasi Perubahan terhadap obat yang telah mendapat nomor ijin edar harus dilaporkan kepada Kepala Badan melalui mekanisme Registrasi variasi. Registrasi variasi adalah registrasi perubahan aspek apapun pada produk yang Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 28 telah memiliki izin edar di Indonesia, termasuk tetapi tidak terbatas pada perubahan formulasi, metoda, proses pembuatan, spesifikasi untuk obat dan bahan baku, wadah, kemasan dan penandaan. Registrasi Variasi Major (VaMa) adalah registrasi variasi yang berpengaruh bermakna terhadap aspek khasiat, keamanan, dan/atau mutu obat. Registrasi Variasi Minor yang Memerlukan Persetujuan (VaMi-B) adalah registrasi variasi yang tidak termasuk kategori registrasi variasi minor dengan notifikasi maupun variasi major. Registrasi Variasi Minor dengan Notifikasi (VaMi-A) adalah registrasi variasi yang berpengaruh minimal atau tidak berpengaruh sama sekali terhadap aspek khasiat, keamanan, dan/atau mutu obat, serta tidak merubah informasi pada sertifikat izin edar (Badan POM RI, 2011). Permohonan registrasi variasi diajukan dengan mengisi formulir dan melampirkan dokumen registrasi variasi terkait perubahan yang diajukan. 4. 5 Registrasi Ulang Registrasi ulang dilakukan pada produk yang telah habis masa nomor ijin edarnya. Permohonan pengajuan registrasi ulang dilakukan paling cepat 120 hari sebelum berakhir masa berlaku izin edar. Permohonan ini diajukan dengan mengisi formulir registrasi dan melampirkan dokumen registrasi ulang. Persetujuan atas permohonan registrasi ulang secara otomatis berlaku sejak berakhir masa izin edarnya, kecuali untuk registrasi ulang dengan informasi terbaru yang terkait aspek keamanan obat, khasiat obat, dan/atau kerasionalan formula obat. 4. 6 Regulatory 4. 6. 1 Fungsi Regulatory Fungsi seorang regulator sangat penting di PT. Novartis Indonesia. Regulator bertanggung jawab untuk meregistrasikan produk yang belum memiliki ijin edar, mengajukan registrasi variasi terhadap perubahan produk yang telah memiliki nomor ijin edar, dan meregistrasi ulang produk yang telah habis masa ijin edarnya. Regulator berperan sebagai perencana proses registrasi, sebagai orang yang mempersiapkan persyaratan registrasi obat meliputi formulir Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 29 maupun dokumen pendukung registrasi obat, sebagai pendaftar ke Badan POM, sebagai pihak yang bernegosiasi jika terjadi permasalahan selama proses registrasi baik dalam bentuk kelengkapan data maupun konsultasi dengan pihak yang terkait, sebagai pihak yang mengurus pembayaran terkait registrasi obat, sebagai pengontrol selama proses registrasi berjalan, dan sebagai pihak yang membuat pelaporan tentang produk-produk yang telah mengalami proses registrasi. 4. 6. 2 Karakterisasi regulator Menjadi seorang regulator harus memiliki beberapa karakter pendukung terkait fungsinya sebagai seorang regulatory. Karakter tersebut antara lain adalah: a. Pintar, artinya seorang regulator dapat menangkap informasi yang terdapat dalam dokumen terkait registrasi dan paham secara keseluruhan isi dokumen tersebut sehingga dapat menghadapi evaluator dari Badan POM dengan baik. b. Memiliki kemampuan dalam mempersiapkan semua persyaratan yang dibutuhkan untuk meregistrasikan produknya, baik terkait dokumen pendukung maupun formulir dan hal-hal lain sehingga memudahkan evaluator memeriksa kelengkapan yang diperlukan dalam meregistrasikan suatu produk. c. Memiliki sikap yang baik dalam bekerja, baik ketika berada dalam suatu tim maupun individual. d. Sikap yang harus dimiliki oleh seorang regulator antara lain disiplin, jujur, teliti, kreatif, mudah bersosialisasi, mudah bekerja sama, dapat bernegosiasi dengan baik, efektif dan efisien dalam mengerjakan suatu pekerjaan. Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 BAB 5 KESIMPULAN DAN SARAN 5.1 Kesimpulan Berdasarkan praktek kerja profesi apoteker yang dilakukan di PT. Novartis, maka dapat disimpulkan bahwa peranan seorang Apoteker di bagian Regualtory Affair di PT. Novartis Indonesia yaitu bertanggung jawab untuk meregistrasikan obat-obatan yang belum memiliki izin edar, melakukan registrasi variasi terhadap perubahan yang dialami oleh obat-obatan yang telah memiliki izin edar dan melakukan registrasi ulang untuk obat-obatan yang telah habis masa izin edarnya. 5.2 Saran a. Pihak Universitas harus mengembangkan kerja sama dengan bagian regulatori industri farmasi dalam pelaksanaan program Praktek Kerja Profesi Apoteker untuk mengembangkan potensi para calon apoteker mengenai tugas dan fungsinya secara lebih luas di Industri Farmasi, tidak hanya terbatas pada bagian Produksi dan Mutu Produk. b. Perlu menanamkan karakter yang harus dimiliki seorang regulator kepada para calon apoteker sehingga dapat memaksimalkan fungsi regulator dalam suatu industri farmasi. 30 Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia DAFTAR PUSTAKA Badan POM RI. (2011). Peraturan Kepala Badan Pengawas Obat dan Makanan Nomor HK.03.1.23.10.11.08481 Tahun 2011 tentang Kriteria dan Tata Laksana Registrasi Obat. Jakarta : Badan Pengawas Obat dan Makanan Republik Indonesia. Novartis Indonesia. (2014). Tentang Novartis Indonesia. Maret 18, 2014. http://www.id.novartis.com. Kementerian Kesehatan RI. (2010). Peraturan Menteri Kesehatan RI No. 1700/MenKes/Per/XII/2010 tentang Industri Farmasi. Jakarta : Kementerian Kesehatan Republik Indonesia. 31 Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia LAMPIRAN Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 33 Lampiran 1. Struktur organisasi Medical and Regulatory Departement, PT. Novartis Indonesia, Jakarta. Chief Scientific Officer (CSO) Secretary CRA Head Medical Affair Manager MA 1 MA 2 MA 3 MIC Manager CRA MSL MSL MSL CRA MSL MSL MSL CRA Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 PV & RMP Manager DRA Head DRA Manage r DRA Officer DRA Staff 34 Lampiran 2. Formulir registrasi Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 35 Lampiran 2. (Lanjutan) Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 36 Lampiran 2. (Lanjutan) Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 37 Lampiran 2. (Lanjutan) Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 38 Lampiran 3. Isi dokumen pra-registrasi dan registrasi BAGIAN I DOKUMEN ADMINISTRATIF DAN INFORMASI PRODUK Sub Bagian A Daftar Isi Keseluruhan Sub Bagian B Dokumen Administratif 1. Surat Pengantar 2. Formulir Registrasi 3. Pernyataan Pendaftar 4. Sertifikat dan Dokumen Administratif Lain 4.1 Obat Produksi Dalam Negeri 4.1.1 Izin industri farmasi 4.1.2 Sertifikat CPOB yang masih berlaku untuk bentuk sediaan yang didaftarkan 4.1.3 Data insperksi terakhir dan perubahan terkait paling lama dua tahun yang dikeluarkan oleh BPOM 4.2 Obat Produksi Dalam Negeri berdasarkan Lisensi 4.2.1 Izin industri farmasi atau dokumen penunjang dengan bukti yang cukup untuk badan atau institusi riset sebagai pemberi lisensi 4.2.2 Izin industri farmasi sebagai penerima lisensi 4.2.3 Sertifikat CPOB industri farmasi penerima lisensi yang masih berlaku untuk bentuk sediaan yang didaftarkan 4.2.4 Perjanjian lisensi 4.3 Obat Produksi Dalam Negeri berdasarkan Kontrak 4.3.1 Izin industri farmasi pendaftar/pemberi kontrak 4.3.2 Izin industri farmasi sebagai penerima kontrak 4.3.2 Sertifikat CPOB industri farmasi pemberi kontrak yang masih berlaku 4.3.3 Perjanjian kontrak 4.4 Obat Khusus Ekspor 4.4.1 Izin industri farmasi 4.4.2 Sertifikat CPOB pendaftar Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 pendaftar atau 39 Lampiran 3. (Lanjutan) 4.4.3 Sertifikat CPOB atau dokumen lain yang setara dari produsen sesuai bentuk sediaan yang didaftarkan (untuk obat impor khusus ekspor) 4.5 Obat Impor 4.5.1 Izin industri farmasi produsen dan pendaftar 4.5.2 Surat penunjukkan dari industri farmasi atau pemilik produk di luar negeri 4.5.3 Certificate of Pharmaceutical Product (CPP) atau dokumen lain yang setara dari negara produsen dan/atau Negara dimana diterbitkan sertifikat kelulusan bets 4.5.4 Sertifikat CPOB yang masih berlaku dari produsen untuk bentuk sediaan yang didaftarkan atau dokumen lain yang setara (termasuk sertifikat CPOB produsen zat aktif untuk produk biologi) 4.5.5 Data inspeksi CPOB terakhir dan perubahan terkait paling lama dua tahun yang dikeluarkan oleh otoritas pengawas obat setempat dan/atau otoritas pengawas obat negara lain 4.5.6 Justifikasi impor 4.5.7 Bukti perimbangan kegiatan ekspor dan impor (jika perlu) 5. Hasil Pra-Registrasi 6. Kuitansi/Bukti Pembayaran 7. Dokumen Lain Sub Bagian C Informasi Produk dan Penandaan 1. Informasi Produk 2. Penandaan pada kemasan BAGIAN II DOKUMEN MUTU Sub Bagian A Ringkasan Dokumen Mutu (RDM) Sub Bagian B Dokumen Mutu S Zat Aktif S1 Informasi Umum Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 40 Lampiran 3. (Lanjutan) S1.1 Tata Nama S1.2 Rumus Kimia S1.3 Sifat-sifat umum S2 Proses Produksi dan Sumber Zat Aktif S2.1 Produsen S2.2 Uraian dan Kontrol Proses Pembuatan S2.3 Kontrol terhadap Bahan S2.4 Kontrol terhadap Tahapan Kritis dan Senyawa S2.5 Validasi proses dan/atau Evaluasi S2.6 Pengembangan Proses Pembuatan S3 Karakterisasi S3.1 Elusidasi dan Struktur Karakterisasi S3.2 Bahan Pengotor S4 Spesifikasi dan Metode Pengujian Zat Aktif S4.1 Spesifikasi antara S4.2 Prosedur Analisis S4.3 Validasi Prosedur Analisis S4.4 Analisis Bets S4.5 Justifikasi Spesifikasi S5 Baku Pembanding S6 Spesifikasi dan Pengujian Kemasan S7 Stabilitas P Obat Jadi P1 Pemerian dan Formula P2 Pengembangan Produk P2.1 Informasi dan Studi Pengembangan P2.2 Komponen Obat P2.2.1 Zat Aktif P2.2.2 Zat Tambahan P2.3 Obat Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 41 Lampiran 3. (Lanjutan) P2.3.1 Pengembangan Formula P2.3.2 Overages P2.3.3 Sifat Fisikokimia dan biologi P2.4 Pengembangan Proses Pembuatan P2.5 Sistem Kemasan P2.6 Atribut Mikrobiologi P2.7 Kompatibilitas P3 Prosedur Pembuatan P3.1 Formula Bets P3.2 Proses Pembuatan dan Kontrol Proses P3.3 Kontrol terhadap Tahapan Kritis dan Produk Antara P3.4 Validasi Proses dan/atau Laporan P4 Spesifikasi dan Metode Pengujian Zat Tambahan P4.1 Spesifikasi P4.2 Prosedur Analisis P4.3 Zat Tambahan yang Bersumber dari Hewan dan/atau Manusia P4.4 Zat Tambahan Baru P5 Spesifikasi dan Metode Pengujian Obat P5.1 Spesifikasi P5.2 Prosedur Analisis P5.3 Laporan validasi Metode Analisis P5.4 Analisis Bets P5.5 Karakterisasi Zat Pengotor P5.6 Justifikasi Spesifikasi P6 Baku Pembanding P7 Spesifikasi dan Metode Pengujian Kemasan P8 Stabilitas P9 Bukti Ekivalensi (bila perlu) Sub Bagian C Daftar Pustaka Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 42 Lampiran 3. (Lanjutan) BAGIAN III DOKUMEN NONKLINIK Sub Bagian A Tinjauan Studi Nonklinik Sub Bagian B Ringkasan dan Matriks Studi Nonklinik Sub Bagian C Laporan Studi Nonklinik Sub Bagian D Daftar Pustaka BAGIAN IV DOKUMEN KLINIK Sub Bagian A Tinjauan Studi Klinik Sub Bagian B Ringkasan Studi Klinik Sub Bagian C Matriks Studi Klinik Sub Bagian D Laporan Studi Klinik Sub Bagian E Daftar Pustaka [Sumber : Badan POM RI, 2011] Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 43 Lampiran 4. Gambar alur proses Tahap pra-registrasi Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 44 Lampiran 5. Gambar alur registrasi Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 45 Lampiran 6. Kelengkapan dokumen registrasi baru No. KATEGORI*) 1 2 1.1 1.2 1.3 1.4 3 1.5 2.1 2.2 BAGIAN I: KELENGKAPAN DOKUMEN ADMINISTRATIF DAN INFORMASI PRODUK A. DOKUMEN ADMINISTRATIF 1 Surat Pengantar v v v v v v v v 2 Formulir Registrasi v v v v v v v v 3 Pernyataan Pendaftar v v v v v v v v 4 Sertifikat dan Dokumen Administratif (sesuai dengan status produksi: obat produksi dalam negeri, kontrak, lisensi, ekspor atau impor) sesuai Lampiran 5. v v v v v v v v 5 Hasil Pra-Registrasi v v v v v v v v 6 Kuitansi/Bukti Pembayaran v v v v v v v v 7 Dokumen Terkait Paten 8 7.1. Surat Pernyataan Terkait Paten va) va) va) va) vb) vb) 7.2. Hasil Penelusuran Paten 7.3. Kajian Mandiri Terkait Paten (self assessment) va) va) va) va) vb) vb) va) va) va) va) vb) vb) Surat pernyataan bermaterai dari produsen v v v v v v v v v v v v v v v v mengenai penggunaan bahan yang bersumber babi / porcine (jika perlu) 9 Surat keterangan dari produsen mengenai penggunaan bahan baku bersumber dari hewan atau bahan baku bersumber dari tumbuhan (termasuk tetapi tidak terbatas pada gelatin; laktosa monohidrat; magnesium stearat; bahan-bahan yang mengandung asam lemak seperti stearat, oleat, palmitat; gliserin dan jenis lemak hidrogenasi; DHA; asam arakhidonat; eudragit) (jika perlu) Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 46 Lampiran 6. (Lanjutan) No. KATEGORI*) 1 2 3 1.1 1.2 1.3 1.4 1.5 2.1 2.2 Jika bersumber dari hewan disertai dengan informasi sumber hewan dan surat keterangan bebas BSE/TSE B. INFORMASI PRODUK DAN PENANDAAN 1 Informasi Produk v v v v v v v V 2 Penandaan v v v v v v v v 3 Contoh obat dan kemasan dalam bentuk foto atau gambar sesuai aslinya (mock up/dummy) v v v v v v v v v v v v v v v v 1.1. Tata Nama v v vc) vc) v v v v 1.2. Rumus Kimia v v vc) vc) v v v v v vc) v v v v BAGIAN II: KELENGKAPAN DOKUMEN MUTU Sub Bagian A. Ringkasan Dokumen Mutu (RDM) Sub. Bagian B. Dokumen Mutu S. ZAT AKTIF S.1. Informasi Umum S.2. 1.3. Sifat-sifat Umum Proses Produksi dan Sumber Zat Aktif 2.1. Produsen 2.2. Uraian dan Kontrol Proses Pembuatan 2.3. Kontrol terhadap bahan 2.4. Kontrol terhadap tahapan kritis dan senyawa antara 2.5. Validasi Proses dan/atau Evaluasi 2.6. Pengembangan Proses Pembuatan v vc) v v v vc) vc) v v v vc) vc) v v v v vc) vc) v v v v vc) vc) v v v v vc) vc) v v v v vc) vc) v v Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 v v v 47 Lampiran 6. (Lanjutan) No. KATEGORI*) 1 2 3 1.1 1.2 1.3 1.4 1.5 2.1 2.2 S.3. Karakterisasi 3.1. Elusidasi dari struktur dan Karakterisasi 3.2. Bahan Pengotor S.4. Spesifikasi dan Metode Pengujian Zat Aktif 4.1. Spesifikasi 4.2. Prosedur Analisis 4.3. Validasi Prosedur Analisis 4.4. Analisis Bets v v v vc) vc) v v v v vc) vc) v v v vc) vc) vc) vc) vc) vc) v v v v v v vc) vc) vc) vc) v vc) v vc) v v vc) vc) v v v vc) vc) v v v v v v v v v v v v v v v v v v v v 2.2. Informasi Studi Pengembangan Komponen Obat v v v v v v v v 2.3. Obat v v v v v v v v v v v 2.5. Pengembangan Proses Pembuatan Sistem Kemasan v v v 2.4. v v v v v v v v 2.6. Atribut Mikrobiologi v v v v v v v v 2.7. Kompatibilitas v v v v v v v v 3.1. Formula Bets v v v v v 3.2. Proses Pembuatan dan Kontrol Proses v v v v v v v v v v v 4.5. Justifikasi Spesifikasi S.5. Baku Pembanding S.6. Spesifikasi dan Pengujian Kemasan S.7. Stabilitas v v v v v v v v v v v v vd) v v v v vd) v v v v v v v v v P. OBAT P.1. Pemerian dan Formula P.2. Pengembangan Produk 2.1. P.3. Prosedur Pembuatan Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 48 Lampiran 6. (Lanjutan) No. KATEGORI*) 1 2 3 1.1 1.2 1.3 1.4 1.5 2.1 2.2 P.4. 3.3. Kontrol terhadap v v v v v v v v Tahapan Kritis dan Produk Antara 3.4. Validasi Proses dan/atau v v v v v v v v v v v v v v v v v v v v v v v v v v v v v v v v v v v v v v v v Laporan Spesifikasi dan Metode Pengujian Zat Tambahan 4.1. Spesifikasi 4.2. Prosedur Analisis 4.3. Zat tambahan bersumber dari Hewan dan/atau Manusia 4.4. Zat Tambahan Baru P.5. Spesifikasi dan Metode Pengujian Obat 5.1. Spesifikasi v v v v v v v v 5.2. Prosedur Analisis v v v v v v v v 5.3. v v v v v v v v 5.4. Laporan Validasi Metode Analisis Analisis Bets v v v v v v v v 5.5. Karakterisasi Zat v v v v v v v v 5.6. Pengotor Justifikasi Spesifikasi v v v v v v v v P.6. Baku pembanding v v v v v v v v P.7. Spesifikasi dan Metode Pengujian Kemasan Stabilitas Bukti Ekivalensi v v v v v v v v v v v v v v v v v v v v v v v v v P.8. P.9 Sub Bagian C. Daftar Pustaka Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 49 Lampiran 6. (Lanjutan) No. KATEGORI*) 1 2 3 1.1 1.2 1.3 1.4 1.5 2.1 2.2 BAGIAN III: KELENGKAPAN DOKUMEN NONKLINIK Sub Bagian A. Tinjauan Studi Nonklinik v v ve) v v vf) Sub Bagian B. Ringkasan dan Matriks Studi Nonklinik v v ve) v v vf) 1 Ringkasan Studi Noklinik v v ve) v v vf) 2 Isi Ringkasan dan Matriks Studi Nonklinik v v ve) v v vf) 3 Ringkasan Matriks Studi Nonklinik v v ve) v v vf) v v ve) v v vf) v v ve) v v vf) 2.1. Farmakologi v v ve) v v vf) 2.2. Farmakokinetik v v ve) v v vf) 2.3. Toksikologi v v ve) v v vf) v v v v v v v v v v v v vg) v Sub Bagian C. Laporan Studi Nonklinik (jika perlu) 1 Daftar Isi Laporan Studi Nonklinik 2 Laporan Studi Sub Bagian D. Daftar Pustaka BAGIAN IV: KELENGKAPAN DOKUMEN KLINIK Sub Bagian A. Tinjauan Studi Klinik v Sub Bagian B. Ringkasan Studi Klinik vg) 1 Ringkasan Studi Biofarmasetika dan Metode Analisis Terkait v v v v v v 2 Ringkasan Studi Farmakologi Klinik v v v v v v 3 Ringkasan Khasiat Klinik v v v v v v 4 Ringkasan Keamanan Klinik v v v v v v 5 Sinopsis Studi Individual v v v v v v Sub Bagian C. Matriks Studi Klinik v v v v v Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 vg) v 50 Lampiran 6. (Lanjutan) No. KATEGORI*) 1 2 3 1.1 1.2 1.3 1.4 1.5 2.1 2.2 Sub Bagian D. Laporan Studi Klinik v v v v v v v v v v v v v v v v v v v v 2.3 Laporan Studi Farmakokinetika (PK) Pada v Manusia 2.3.1. Laporan Studi PK pada Subyek Sehat dan Tolerabilitas Awal 2.3.2. Laporan Studi PK pada Subyek dan Laporan Tolerabilitas Awal 2.3.3. Laporan Studi PK pada Populasi v v v v v 2.4 Laporan Studi Farmakodinamika (PD) Pada Manusia 2.4.1. Laporan Studi PD dan PK/ PD pada Subyek Sehat 2.4.2. Laporan Studi PD dan PK/ PD pada Subyek v v v v v 1 Daftar Isi Laporan Studi Klinik 2 Laporan Studi Klinik 2.1 Laporan Studi Biofarmasetika 2.1.1. 2.1.2. 2.1.3. 2.1.4. vg) v v v Laporan Studi Ketersediaan Hayati / Bioavailability (BA) Laporan Studi Perbandingan Ketersediaan Hayati / Bioavailability (BA) dan Bioekivalensi (BE) Laporan Studi Korelasi In vitro-In vivo Laporan Metode Bioanalisis dan Analisis untuk Studi pada Manusia 2.2 Laporan Studi terkait Farmakokinetik Menggunakan Biomaterial Manusia 2.2.1 Laporan Studi Ikatan Protein Plasma 2.2.2. Laporan Studi Metabolisme Hati dan Interaksi Obat 2.2.3. Laporan Studi Menggunakan Biomaterial Manusia Lainnya v Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 51 Lampiran 6. (Lanjutan) No. KATEGORI*) 1 2 3 1.1 1.2 1.3 1.4 1.5 2.1 2.2 2.5 Laporan Studi Khasiat dan Keamanan 2.5.1. 2.5.2. 2.5.3. 2.5.4. v v v v v v Laporan Studi Klinik Berpembanding Terkait Klim Indikasi Laporan Studi Klinik Tanpa Pembanding Laporan Analisis Data dari Lebih dari Satu Studi, Termasuk Analisis Formal Terpadu, Metaanalisis, dan Bridging Analysis. Laporan Studi Klinik Lain 3 Laporan Pengalaman Paska Pemasaran v v v v v v 4 Formulir Laporan Kasus dan Daftar Subyek Individual (jika perlu) v v v v v v v v v v v Sub Bagian E. Daftar Pustaka vg) Keterangan : a) V : jika pendaftar bukan inovator atau tidak mendapat penunjukan/lisensi dari inovator b) V : untuk obat copy pertama c) V : jika sumber zat aktif berbeda dari yang disetujui ) Vd : untuk zat aktif non kompendial e) V : untuk rute pemberian baru f) V : dipersyaratkan untuk komponen obat yang belum pernah disetujui g) V : untuk obat copy yang memerlukan uji klinik *) : registrasi obat impor bentuk ruahan yang akan dikemas kembali dan diedarkan di Indonesia didaftarkan bersamaan dengan kemasan siap edarnya [Sumber : Badan POM RI, 2011] Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 v 52 Lampiran 7. Dokumen registrasi variasi Dokumen administratif yang harus diserahkan pada saat pengajuan registrasi variasi meliputi: 1. Surat Penngantar. 2. Formulir Registrasi. 3. Pernyataan Pendaftar. 4. Sertifikat dan Dokumen Administratif (sesuai dengan status produksi: obatproduksi dalam negeri, kontrak, lisensi, ekspor, impor) sesuai Lampiran 5. 5. Hasil Pra Registrasi (jika dipersyaratkan). 6. Kuitansi/Bukti Pembayaran. 7. Dokumen lain-lain. 7.1.Surat pernyataan terkait pemenuhan persyaratan registrasi variasi (misal: surat pernyataan bahwa prosedur pengujian zat aktif tidak berubah untuk registrasi variasi pengetatan batas spesifikasi zat aktif). 7.2.Copy NIE dan semua surat persetujuan perubahan yang diterbitkan oleh Badan POM beserta lampirannya. 7.3.Tabel sandingan perubahan yang diajukan, termasuk referensi perubahan. 7.4. Justifikasi terhadap perubahan yang diajukan. [Sumber : Anonim, 2011] Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 UNIVERSITAS INDONESIA TUGAS KHUSUS PRAKTEK KERJA PROFESI APOTEKER DI PT. NOVARTIS INDONESIA JL. PROF. DR. SATRIO KAV. 18 KUNINGAN CITY SETIABUDI JAKARTA SELATAN 12940 PERIODE 4 FEBRUARI - 28 MARET 2014 PENYUSUNAN DAN PROSES PENGAJUAN DOKUMEN REGISTRASI VARIASI TERKAIT PENANDAAN PRODUK B PT. NOVARTIS INDONESIA KE BADAN POM ASVINASTUTI RIKASIH, S. Farm. 1306343416 ANGKATAN LXXVIII FAKULTAS FARMASI PROGRAM PROFESI APOTEKER DEPOK JUNI 2014 Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 UNIVERSITAS INDONESIA PENYUSUNAN DAN PROSES PENGAJUAN DOKUMEN REGISTRASI VARIASI TERKAIT PENANDAAN PRODUK B PT. NOVARTIS INDONESIA KE BADAN POM Diajukan sebagai salah satu syarat untuk memperoleh gelar Apoteker ASVINASTUTI RIKASIH, S. Farm. 1306343416 ANGKATAN LXXVIII FAKULTAS FARMASI PROGRAM PROFESI APOTEKER DEPOK JUNI 2014 ii Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia DAFTAR ISI HALAMAN JUDUL .......................................................................................... DAFTAR ISI ...................................................................................................... DAFTAR GAMBAR .......................................................................................... DAFTAR TABEL ............................................................................................... DAFTAR LAMPIRAN ....................................................................................... ii iii iv v vi BAB 1. PENDAHULUAN ................................................................................. 1 1.1. Latar Belakang ............................................................................... 1 1.2. Tujuan ............................................................................................ 2 BAB 2. TINJAUAN PUSTAKA ....................................................................... 2.1. Registrasi ........................................................................................ 2.2. Tata Laksana Registrasi Obat .......................................................... 2.3. Dokumen Registrasi ....................................................................... 2.4. ASEAN Common Technical Dossier / ASEAN Common Technical requirements ................................................................................... 2.5. Registrasi Variasi ............................................................................ 2.6. Perubahan Penandaan Produk PT. Novartis Indonesia .................... 2.7. Produk B PT. Novartis Indonesia .................................................... 3 3 4 6 7 9 15 18 BAB 3. METODOLOGI .................................................................................... 3. 1 Waktu ............................................................................................. 3. 2 Lokasi ............................................................................................ 3. 3 Data ................................................................................................ 3. 4 Metode .......................................................................................... 20 20 20 20 20 BAB 4. HASIL DAN PEMBAHASAN .............................................................. 4. 1 Informasi perubahan dari Novartis global ke DRA Novartis Indonesia ........................................................................................ 4. 2 Pengambilan dokumen dari sistem informasi terkait perubahan penandaan produk B ....................................................................... 4. 3 Penyusun dossier dan pengecekan kelengkapan dokumen ............... 4. 4 Proses submit atau pengajuan dokumen di BPOM ........................... 22 23 24 25 27 BAB 5. KESIMPULAN DAN SARAN .............................................................. 30 5. 1 Kesimpulan ..................................................................................... 30 5. 2 Saran .............................................................................................. 30 DAFTAR ACUAN ............................................................................................ 31 iii Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia DAFTAR GAMBAR Gambar 2.1 Alur Registrasi ........................................................................ 6 Gambar 2.2 Alur perubahan penandaan terkait safety update mulai dari pemberitahuan awal sampai pembuatan CRPM ....................... 18 iv Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia DAFTAR TABEL Tabel 2.1 SKU Produk B PT. Novartis Indonesia ..................................... 19 v Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia DAFTAR LAMPIRAN Lampiran 1 Formulir Registrasi Obat dan Produk Biologi ........................... Lampiran 2 Dokumen Administratif Registrasi Variasi ............................... Lampiran 3 Informasi Minimal yang Harus Dicantumkan Pada Penandaan ............................................................................... Lampiran 4 Hasil Pra-Registrasi (HPR) Produk B ...................................... Lampiran 5 Cover ordner Produk B ............................................................ Lampiran 6 Contoh Checklist registrasi Variasi Major (VaMa) Produk B dan cara pengisiannya ............................................................... Lampiran 7 Proses submit atau pengajuan dokumen registrasi variasi di Badan POM .............................................................................. vi Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 33 37 38 40 41 42 45 Universitas Indonesia 1 BAB 1 PENDAHULUAN 1.1 Latar Belakang Obat adalah obat jadi termasuk produk biologi, yang merupakan bahan atau paduan bahan yang digunakan untuk mempengaruhi atau menyelidiki sistem fisiologi atau keadaan patologi dalam rangka penetapan diagnosis, pencegahan, penyembuhan, pemulihan dan peningkatan kesehatan dan kontrasepsi untuk manusia. Obat jadi yang diproduksi oleh industri farmasi harus memenuhi persyaratan Cara Pembuatan Obat yang Baik (CPOB) selama masa produksi dan harus memiliki izin edar sebelum diedarkan di Indonesia yang diperoleh melalui proses registrasi kepada Kepala Badan Pengawasan Obat dan Makanan (BPOM). Proses registrasi bertujuan untuk memberikan perlindungan kepada masyarakat dari peredaran obat yang tidak memenuhi persyaratan khasiat, keamanan, dan mutu (Badan POM RI, 2011). Produk obat yang telah memiliki izin edar dapat mengalami perubahan berkaitan dengan dilakukannya pengembangan atau pembaruan. Perubahan yang dilakukan dapat berkaitan dengan aspek khasiat, keamanan, dan/ atau mutu obat maupun perubahan minimal yang tidak berpengaruh terhadap aspek khasiat, keamanan, dan/ atau mutu obat. Segala jenis perubahan yang terjadi pada obat harus dilaporkan ke Badan POM melalui suatu prosedur mekanime registrasi variasi yang pelaksaannya berpedoman pada Peraturan Kepala BPOM RI No. HK.03.1.23.10.11.08481 tahun 2011 tentang Kriteria dan Tata Laksana Registrasi Obat (Buku Coklat). Hal ini dimaksudkan agar setiap obat yang beredar di pasaran dan dikonsumsi oleh masyarakat adalah obat yang memiliki khasiat, keamanan, dan mutu yang sama dengan obat yang telah terdaftar di Badan POM (Badan POM RI, 2011). PT. Novartis Indonesia merupakan salah satu anak perusahaan dari perusahaan Novartis International AG, yaitu perusahaan farmasi multinasional berbasis riset yang berlokasi di Basel, Swiss (Novartis Indonesia, 2014). PT. Novartis Indonesia selalu melakukan pengembangan melalui riset dan evaluasi terhadap produk-produk obatnya yang telah beredar di pasaran. Perubahan yang 1 Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia 2 dilakukan pada produk obatnya, akan didaftarkan oleh regulator PT. Novartis Indonesia, yaitu bagian Drug Regulatory Affair (DRA) sesuai prosedur registrasi variasi. Di bagian DRA PT. Novartis Indonesia, perubahan yang ada dibagi menjadi dua kategori, yaitu perubahan yang terkait mutu (quality update) dan juga perubahan yang terkait dengan penandaan (safety update) produk. Tugas khusus ini membahas tentang registrasi variasi yang berhubungan dengan adanya perubahan terkait penandaan (safety update) pada produk dari PT. Novartis Indonesia, khususnya proses penyusunan dokumen serta proses pengajuannya kepada BPOM. Penyusunan dokumen dan pengajuan registrasi variasi harus sesuai dengan pedoman Peraturan Tata laksana registrasi obat yang diatur oleh Badan POM. Oleh karena itu, dilakukan kerjasama antara Program Profesi Apoteker Fakultas Farmasi Universitas Indonesia dengan PT. Novartis Indonesia melalui program Praktek Kerja Profesi Apoteker (PKPA) yang dilaksanakan pada 4 Februari 2014 hingga 28 Maret 2014 untuk memberikan pengetahuan dan pengalaman bagi calon apoteker mengenai registrasi variasi, khususnya mengenai penyusunan dokumen registrasi variasi yang benar dan proses pengajuannya. Selain itu, untuk melatih calon apoteker agar nantinya dapat bekerja secara profesional, khususnya sebagai regulator yang memiliki ketelitian dalam penyusunan dokumen registrasi, dapat menangkap informasi yang ada dalam dokumen registrasi, serta memiliki kemampuan berkomunikasi pada saat proses evaluasi dengan evaluator BPOM. 1.1 Tujuan Mengetahui, memahami, dan mampu menyusun dokumen registrasi variasi produk B PT. Novartis Indonesia, serta dapat melakukan proses pengajuan dokumennya ke BPOM sesuai dengan Peraturan Tata laksana registrasi obat yang diatur oleh Badan POM dalam keputusan Kepala Badan Pengawas Obat dan Makanan Republik Indonesia Nomor HK.03.1.23.10.11.08481 Tahun 2011 Tentang Kriteria dan Tata Laksana Registrasi Obat. Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 3 BAB 2 TINJAUAN UMUM 2. 1 Registrasi 2. 2. 1 Definisi Registrasi Registrasi adalah prosedur pendaftaran dan evaluasi obat untuk mendapatkan izin edar. Izin edar adalah bentuk persetujuan registrasi obat untuk dapat diedarkan di wilayah Indonesia. Kriteria obat yang dapat memiliki izin edar antara lain (Kemenkes, 2008): a. Khasiat yang meyakinkan dan keamanan yang memadai dibuktiin melalui percobaan hewan dan uji klinik atau bukti-bukti lain sesuai dengan status perkembangan ilmu pengetahuan yang bersangkutan. b. Mutu yang memenuhi syarat yang dinilai dari proses produksi sesuai Cara Pembuatan Obat Yang Baik (CPOB), spesifikasi dan metode pengujian terhadap semua bahan yang digunakan serta produk jadi dengan bukti yang sahih. c. Penandaan dan informasi produk berisi informasi lengkap, obyektif, dan tidak menyesatkan yang dapat menjamin penggunaan obat secara tepat, rasional, dan aman. d. Sesuai dengan kebutuhan nyata masyarakat. e. Khusus untuk psikotropika baru harus memiliki keunggulan dibandingkan dengan obat yang telah disetujui beredar di Indonesia. f. Khusus untuk kontrasepsi atau obat lain yang digunakan dalam program nasional, dipersyaratkan harus uji klinik di Indonesia. Registrasi obat dilaksanakan di Direktorat Penilaian Obat dan Produk Biologi Badan POM, diajukan kepada Kepala BPOM oleh pendaftar. Tujuan dilakukannya registrasi yaitu untuk melindungi masyarakat dari peredaran obat yang tidak memenuhi persyaratan khasiat, keamanan, mutu, dan manfaatnya. Untuk tujuan ini, maka semua obat yang diedarkan di wilayah Indonesia, sebelumnya harus dilakukan registrasi, kecuali untuk obat dengan beberapa kriteria khusus seperti obat untuk donasi, obat untuk uji klinik, obat sampel untuk 3 Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia 4 registrasi, dan obat dengan penggunaan khusus atas permintaan dokter. Obat ini dapat dimasukkan ke wilayah Indonesia melalui mekanisme jalur khusus yang telah ditetapkan oleh Menteri Kesehatan. 2. 2. 2 Kategori Registrasi Registrasi terdiri atas kategori registrasi baru, registrasi variasi, dan registrasi ulang. 2. 2. 2. 1 Registrasi Baru Registrasi baru merupakan registrasi yang diperuntukkan untuk produk yang belum memiliki izin edar. Registrasi baru terdiri atas Kategori 1 untuk Registrasi Obat Baru dan Produk Biologi, termasuk Produk Biologi Sejenis (PBS)/ Similar Biotherapeutic Product (SBP), sedangkan Kategori 2 untuk registrasi Obat Copy (Badan POM RI, 2011). 2. 2. 2. 2 Registrasi Variasi Registrasi variasi adalah registrasi yang diperuntukkan untuk produk yang telah mempunyai izin edar yang mengalami perubahan. Kategori registrasi variasi terdiri dari kategori 4 untuk registrasi variasi major (VaMa), kategori 5 untuk registrasi variasi minor yang memerlukan persetujuan (VaMi-B), dan kategori 6 untuk registrasi variasi minor dengan notifikasi (VaMi-A) (Badan POM RI, 2011). 2. 2. 2. 3 Registrasi Ulang Registrasi ulang adalah registrasi yang diperuntukkan untuk produk yang mempunyai izin edar yang telah habis masa berlakunya (5 tahun). Registrasi ini termasuk kategori 7 (Badan POM RI, 2011). 2. 2 Tata Laksana Registrasi Obat Permohonan pra-registrasi dan registrasi diajukan oleh pendaftar secara tertulis kepada Kepala BPOM dengan melampirkan dokumen pra-registrasi atau dokumen registrasi. Proses registrasi dibagi ke dalam dua tahap, yaitu tahap praregistrasi dan tahap registrasi. Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 5 a. Tahap Pra-registrasi Permohonan pra-registrasi dilakukan untuk penapisan registrasi obat, penentuan kategori registrasi, penentuan jalur evaluasi, penentuan biaya evaluasi, dan penentuan dokumen registrasi obat. Proses pra-registrasi hanya dilakukan untuk registrasi obat dengan kategori registrasi baru dan registrasi variasi major (VaMa). Untuk kategori registrasi minor dan registrasi ulang tidak dipersyaratkan melakukan pra-registrasi (Badan POM RI, 2011). Permohonan pra-registrasi diajukan dengan mengisi formulir pra-registrasi, menyerahkan bukti pembayaran biaya pra-registrasi, dan melampirkan dokumen lengkap pra-registrasi. Pada tahap ini, paling lama dalam jangka waktu 40 hari sejak diterimanya permohonan praregistrasi, Kepala BPOM memberikan surat Hasil Pra-Registrasi (HPR) kepada pendaftar yang berlaku satu tahun sejak tanggal dikeluarkan. b. Tahap Registrasi Pengajuan registrasi dilakukan dengan menyerahkan berkas registrasi dengan mengisi formulir registrasi (Lampiran 1) dan disket disertai bukti pembayaran biaya evaluasi dan pendaftaran, serta HPR bila ada. Berkas registrasi terdiri atas formulir registrasi dengan dokumen administratif dan dokumen penunjang. Dokumen tersebut disusun sesuai format ASEAN Common Technical Dossier (ACTD). Dossier adalah kumpulan dokumen registrasi suatu produk untuk diajukan ke Badan POM sebagai bagian dari persyaratan registrasi (Badan POM RI, 2011). Setelah dilakukan penyusunan dokumen registrasi sesuai dengan persyaratan BPOM, pendaftar mengajukan permohonan registrasi. Dokumen registrasi akan dievaluasi kelengkapannya di loket registrasi oleh evaluator di BPOM setelah permohonan registrasi diterima. Jika tidak lengkap, maka dokumen akan dikembalikan kepada pendaftar untuk dilengkapi. Sedangkan jika sudah lengkap, maka pendaftar akan mendapatkan Surat Perintah Bayar (SPB). Setelah membayar, pendaftar menyerahkan bukti bayar ke loket registrasi, kemudian akan diberikan nomor pengisian formulir elektronik. Pendaftar mengisi formulir elektronik pada komputer yang ada di BPOM, kemudian menyerahkan dokumen registrasi dalam bentuk dossier ke loket penyerahan dokumen BPOM. Pendaftar akan mendapatkan nomor FERO, yaitu nomor identitas registrasi untuk Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 6 melakukan penelusuran proses evaluasi terhadap produk yang diregistrasikannya. Dokumen selanjutnya akan di evaluasi oleh evaluator BPOM sesuai dengan jalur evaluasinya. Dibawah ini adalah bagan dari alur registrasi. [Sumber : Badan POM RI, 2011, telah diolah kembali] Gambar 2.1 Alur Registrasi 2. 3 Dokumen Registrasi Dokumen registrasi adalah berkas registrasi terdiri dari dokumen-dokumen yang disusun menurut halaman dan penomoran yang berurutan, serta setiap dokumen dipisahkan oleh kertas pembatas. Dokumen registrasi merupakan dokumen rahasia yang dipergunakan hanya untuk keperluan evaluasi oleh yang berwenang. Dokumen registrasi terdiri atas : a. Bagian I : Dokumen Administratif, Informasi Produk, dan Penandaan b. Bagian II : Dokumen Mutu c. Bagian III : Dokumen Non-Klinik d. Bagian IV : Dokumen klinik Selain untuk registrasi baru, pedoman penyusunan dokumen registrasi dapat juga untuk penyusunan tambahan data. Setiap bagian dokumen registrasi harus disatukan dalam ordner atau map terpisah atau beberapa bagian dokumen registrasi dapat digabungkan dalam ordner dengan kertas pembatas diantara setiap Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 7 dokumen tersebut. Penggunaan ordner atau map disesuaikan dengan banyaknya dokumen registrasi. Dokumen registrasi yang diserahkan harus dilengkapi dengan rancangan kemasan dan brosur. Rancangan kemasan, meliputi etiket, dus atau bungkus luar, strip atau blister, catch over, ampul atau vial, dan kemasan lain sesuai ketentuan tentang pembungkusan luar dan penandaan yang berlaku, yang merupakan rancangan kemasan obat yang akan diedarkan, dan dilengkapi dengan rancangan warna. Dokumen registrasi variasi dapat dilihat pada Lampiran 2. 2. 4 ASEAN Common Technical Dossier / ASEAN Common Technical Requirements ASEAN Common Technical Dossier (ACTD) adalah format umum yang digunakan untuk menyusun dokumen registrasi yang akan didaftarkan kepada badan regulasi obat di wilayah ASEAN. Tujuan penggunaan ACTD adalah agar penggambaran berbagai informasi produk menjadi transparan dan tidak ambigu, sehingga mempermudah pemeriksaan data-data dasar dan membantu pembaca menjadi lebih cepat terorientasi kepada isi pendaftaran produk. ASEAN Common Technical Requirements (ACTR) adalah seluruh materi tertulis yang bertujuan untuk membantu pendaftar untuk menyiapkan dokumen registrasi secara konsisten sesuai dengan harapan seluruh badan otoritas regulasi obat di ASEAN. ACTR ini berisi seluruh persyaratan dan parameter-parameter yang harus dipenuhi oleh produk obat, baik dari segi kualitas, keamanan, dan efikasi. Untuk memenuhi persyaratan yang tercantum dalam ACTR, maka dibuat berbagai pedoman, seperti pedoman uji stabilitas, validasi analisis, validasi proses, validasi uji bioekivalensi, dan berbagai pedoman keamanan dan pedoman efikasi. 2. 4. 1 Ketentuan ACTD Teks dan tabel harus disiapkan dengan margin yang memungkinkan dokumen dapat dicetak dengan baik pada kertas berukuran A4. Margin kiri sebaiknya cukup besar sehingga informasi tidak bias dengan menggunakan metode binding. Jenis huruf dan besar huruf adalah Times New Roman 12. Untuk teks dan tabel, sebaiknya jenis dan ukuran huruf yang cukup besar sehingga mudah dibaca bahkan setelah difotokopi. Setiap halaman harus diberi angka dengan halaman pertama pada setiap bagian disebut sebagai halaman 1. Untuk Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 8 singkatan dan pengertian Common Technical Dossier harus dijelaskan setiap pertama kali digunakan pada setiap bagian. Referensi harus dicantumkan menurut 1979 Vancouver Declaration of Uniform requirements for Manuscript Submitted to Biomedical Journals. 2. 4. 2 Pembagian ACTD ACTD dibagi ke dalam 4 bagian, yaiu Dokumen Administratif, Dokumen Mutu, Dokumen Non klinik, dan Dokumen Klinik. a. Dokumen Administratif Dokumen ini berisi pengenalan umum dari sediaan farmasi yang akan didaftarkan. Pada bagian awal dokumen ini berisi keseluruhan tabel isi atau keseluruhan dokumen ACTD untuk memberikan informasi dasar yang dapat dicari langsung. Selanjutnya, dokumen ini berisi data administratif yang memerlukan dokumentasi spesifik sejelas mungkin, yaitu formulir registrasi, label, brosur, kemasan, dan lain-lain. Bagian akhir dari dokumen ini adalah informasi produk yang memberikan informasi seperlunya, meliputi informasi pemberian obat, mekanisme kerja, efek samping, dan sebagainya. b. Dokumen Mutu Bagian ini berisikan penjelasan mengenai kualitas produk obat secara menyeluruh beserta laporan penelitiannya. Dokumen kontrol kualitas harus dijelaskan sejelas mungkin. c. Dokumen Non klinik Bagian ini harus memberikan penjelasan non klinik yang disertai dengan rangkuman non klinik tertulis dan rangkuman non klinik dalam bentuk tabel. Data dalam bagian ini tidak disertakan pada pendaftaran produk generik, produk yang telah memiliki NIE dengan variasi minor, dan juga pada beberapa produk dengan variasi mayor. Untuk negara-negara anggota ASEAN tertentu, laporan penelitian dari bagian ini mungkin tidak dibutuhkan untuk produk dengan zat kimia baru (New Chemical Entity/NCE), produk bioteknologi, dan produk dengan variasi mayor lain jika produk aslinya telah teregistrasi dan sudah disetujui untuk izin pemasaran di negara asalnya. Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 9 d. Dokumen Klinik Bagian ini memberikan penjelasan klinik dan rangkuman klinik. Dokumen dari bagian ini juga tidak disertakan pada pendaftaran produk generik, produk yang telah memiliki NIE dengan variasi minor, dan juga pada beberapa produk dengan variasi mayor. Untuk negara-negara anggota ASEAN, laporan penelitian dari bagian ini mungkin tidak dibutuhkan untuk produk dengan zat kimia baru (NCE), produk bioteknologi, dan produk dengan variasi mayor lain jika produk aslinya telah teregistrasi dan sudah disetujui untuk izin pemasaran di Negara asalnya. 2. 5 Registrasi Variasi Registrasi variasi dilakukan apabila terjadi perubahan terhadap obat yang telah mendapat NIE. Perubahan terhadap obat yang telah mendapatkan izin edar harus dilaporkan kepada Kepala Badan melalui mekanisme Registrasi Variasi. Permohonan registrasi variasi diajukan dengan mengisi formulir sebagaimana yang tertera pada Lampiran 1, serta membawa kelengkapan dokumen Registrasi Variasi (Lampiran 2) terkait perubahan yang diajukan sesuai dengan pedoman registrasi yang berlaku yakni Keputusan Kepala Badan Pengawas Obat dan Makanan Republik Indonesia Nomor HK.03.1.23.10.11.08481 Tahun 2011 Tentang Kriteria dan Tata Laksana Registrasi Obat (Buku Coklat). Registrasi variasi terdiri atas tiga kategori, yaitu kategori 4 (Registrasi variasi major), kategori 5 (Registrasi variasi minor yang memerlukan persetujuan), dan kategori 6 (Registrasi variasi minor dengan notifikasi). 2. 5. 1 Registrasi variasi major (VaMa) Suatu perubahan dikategorikan sebagai VaMa apabila perubahan yang terjadi bermakna terhadap khasiat, keamanan dan mutu obat. Perubahan – perubahan tersebut mencakup perubahan informasi produk yang mempengaruhi aspek khasiat keamanan yang memerlukan data uji klinik, perubahan terkait zat aktif dan/atau formula yang mempengaruhi aspek khasiat-keamanan yang memerlukan data uji klinik, perubahan informasi produk yang mempengaruhi aspek keamanan yang tidak memerlukan data uji klinik, perubahan terkait mutu zat aktif dan obat jadi. Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 10 2. 5. 2. 1 Perubahan Informasi Produk yang Mempengaruhi Aspek Khasiat Keamanan yang Memerlukan Data Uji Klinik. a. Perubahan indikasi dan/atau posologi; penambahan indikasi dan/atau posologi baru. b. Perubahan informasi produk yang mempengaruhi aspek keamanan 2. 5. 2. 2 Perubahan terkait zat aktif dan/atau formula yang mempengaruhi aspek khasiat-keamanan yang memerlukan data uji klinik a. Perubahan terkait zat aktif dan/atau formula yang memerlukan uji klinik b. Penggantian Master Cell/Seed Bank 2. 5. 2. 3 Perubahan informasi produk yang mempengaruhi aspek keamanan yang tidak memerlukan data uji klinik a. Perubahan informasi produk yang mempengaruhi aspek keamanan 2. 5. 2. 4 Perubahan terkait mutu zat aktif a. Perubahan dan/atau penambahan produsen zat aktif b. Perubahan proses pembuatan zat aktif atau bahan awal/produk antara zat aktif c. Perubahan spesifikasi IPC dalam proses pembuatan zat aktif. d. Perubahan spesifikasi zat aktif non farmakope e. Perubahan spesifikasi release dan shelf-life zat aktif. f. Perubahan zat tambahan pada zat aktif produk biologi g. Perubahan prosedur pengujian IPC, release dan stabilitas zat aktif h. Perubahan sistem kemasan zat aktif i. Penambahan/update/perubahan pada Plasma Master File 2. 5. 2. 5 Perubahan terkait mutu obat jadi a. Peningkatan ukuran bets obat lebih dari 10 kali b. Peningkatan ukuran bets obat hingga 10 kali, untuk produk steril c. Penurunan ukuran bets obat hingga 10 kali, untuk produk steril d. Perubahan berat penyalut tablet atau berat cangkang kapsul sediaan gastroresistant, modifikasinya atau sediaan lepas lambat e. Perubahan kuantitatif dan/atau kualitatif zat tambahan f. Perubahan zat tambahan produk biologi Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 11 g. Perubahan proses produksi obat yang dapat mempengaruhi stabilitas h. Perubahan proses pembuatan obat jadi di produsen obat jadi yang sama i. Perubahan tempat sebagian atau keseluruhan tahapan produksi obat j. Perubahan tempat pengemasan primer obat k. Perubahan spesifikasi obat non Farmakope l. Perubahan bentuk dan/atau dimensi kemasan primer (untuk sediaan steril) m. Perubahan spesifikasi release dan shelf-life obat. n. Perubahan spesifikasi IPC dalam proses pembuatan obat. o. Perubahan prosedur pengujian zat tambahan pada obat p. Perubahan prosedur pengujian IPC dalam proses pembuatan obat q. Perubahan prosedur pengujian obat untuk pelulusan/studi stabilitas r. Perubahan sistem kemasan obat s. Perubahan sistem kemasan pelarut 2. 5. 2 Registrasi variasi minor yang memerlukan persetujuan (VaMi-B) Suatu perubahan dikategorikan sebagai VaMi-B apabila perubahan yang terjadi tidak termasuk ke dalam kategori registrasi variasi mayor ataupun registrasi variasi minor dengan notifikasi. Jenis perubahan yang tercakup dalam VaMi-B diantaranya adalah perubahan terkait informasi produk, mutu zat aktif serta mutu obat jadi. 2. 5. 2. 1 Perubahan terkait infomasi produk dan/atau penandaan a. Perubahan informasi produk b. Perubahan nama pendaftar / industri farmasi/ pemberi lisensi/ industri farmasi sebagai sumber impor obat c. Perubahan nama dagang obat d. Perubahan informasi produk dan/atau penandaan berdasarkan keputusan pemerintah e. Penambahan besar kemasan f. Penambahan informasi produk dalam bahasa Inggris /Indonesia g. Pengetatan klim yang berkaitan dengan keamanan Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 12 2. 5. 2. 2 Perubahan terkait mutu zat aktif a. Perubahan minor pada proses pembuatan zat aktif b. Perubahan minor pada proses pembuatan zat aktif c. Perubahan metode analisis zat aktif (nonkompendial) d. Penambahan atau perubahan tempat pengujian zat aktif termasuk pengujian untuk studi stabilitas dan IPC e. Perubahan periode uji ulang/shelf life zat aktif 2. 5. 2. 3 Perubahan terkait mutu obat jadi a. Perubahan industri penanggung jawab pelulusan bets (tidak termasuk pengujian obat) b. Perubahan industri penanggung jawab pelulusan bets (termasuk pengujian obat) c. Peningkatan dan/atau penurunan ukuran bets obat hingga 10 kali, untuk bentuk sediaan tablet biasa dan cairan oral d. Perubahan spesifikasi zat tambahan non Farmakope untuk memenuhi persyaratan Farmakope e. Perubahan produsen cangkang kapsul f. Perubahan ukuran cangkang kapsul g. Bentuk atau Dimensi tablet gastro-resistant, tablet lepas lambat, dan scored tablet h. Bentuk atau Dimensi tablet lepas cepat, kapsul, supositoria atau pesari i. Perubahan metode analisis obat j. Perubahan bentuk dan/atau dimensi kemasan primer (untuk sediaan non steril) k. Perubahan besar volume sediaan nonparenteral multi dosis l. Perubahan bahan kemasan sekunder m. Penambahan lokasi pengujian stabilitas n. Perubahan kondisi penyimpanan obat, termasuk produk yang direkonstitusi o. Perpanjangan batas kadaluarsa obat : Kemasan belum dibuka p. Perpanjangan batas kadaluarsa obat : Setelah kemasan dibuka atau setelah rekonstitusi q. Perubahan minor pada proses pembuatan obat jadi Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 13 2. 5. 3 Registrasi variasi minor dengan notifikasi (VaMi-A) Suatu perubahan dikategorikan sebagai VaMi-A apabila perubahan yang terjadi berdapak minimal ataupun tidak sama sekali terhadap khasiat, keamanan, & mutu obat. Jenis perubahan yang tercakup dalam VaMi-A diantaranya adalah perubahan terkait informasi produk, mutu zat aktif serta mutu obat jadi. 2. 5. 3. 1 Perubahan terkait informasi produk dan/atau penandaan a. Perubahan atau penambahan logo (termasuk logo perusahaan) b. Penambahan klim efek samping dan/atau kontra indikasi pada informasi produk c. Pengurangan tempat produksi (termasuk zat aktif, produk antara atau obat, lokasi pengemasan, tempat pelulusan bets) d. Perubahan nama zat aktif e. Perubahan pada bagian dari kemasan primer yang tidak kontak dengan obat (seperti warna flip-off caps, warna ring pada ampul, perubahan pada pelindung jarum (digunakan plastic yang berbeda)) f. Penghilangan bahasa asing dari penandaan obat g. Perubahan bentuk dan/atau dimensi kemasan h. Perubahan desain Kemasan i. Perubahan alamat (redaksional) pendaftar/industry farmasi/pemberi lisensi j. Perubahan system penomoran bets 2. 5. 3. 2 Perubahan terkait mutu zat aktif a. Perubahan nama dan/atau alamat produsen zat aktif b. Update Ph. Eur. Certificate of Suitability (CEP) c. Perubahan pencantuman edisi farmakope untuk zat aktif d. Pengetatan batas spesifikasi zat aktif e. Perubahan spesifikasi zat aktif untuk memenuhi persyaratan Farmakope terbaru f. Perubahan spesifikasi zat aktif non Farmakope untuk memenuhi persyaratan Farmakope Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 14 g. Penambahan parameter pengujian dan batas spesifikasi IPC dalam proses pembuatan zat aktif h. Perubahan minor pada prosedur analisis zat aktif i. Perubahan metode analisis penetapan kadar zat aktif sesuai dengan monografi Farmakope j. Perubahan kondisi penyimpanan zat aktif k. Perubahan Working Cell/Seed Bank 2. 5. 3. 3 Perubahan terkait mutu obat jadi a. Perubahan minor pada pembuatan obat b. Perubahan atau penambahan tempat pengujian obat c. Pengetatan batas spesifikasi pelulusan obat d. Penambahan parameter pengujian dan batas spesifikasi IPC dalam proses pembuatan obat e. Pengetatan batas spesifikasi inprocess selama pembuatan obat f. Penambahan parameter pengujian obat g. Pengetatan batas spesifikasi zat tambahan h. Perubahan minor pada prosedur analisis zat tambahan i. Perubahan prosedur analisis zat tambahan sesuai dengan monografi farmakope atau yang relevan j. Penambahan parameter uji pada spesifikasi zat tambahan k. Perubahan pada prosedur analisis zat tambahan, termasuk penggantian metode pengujian l. Perubahan spesifikasi zat tambahan untuk memenuhi persyaratan Farmakope m. Perubahan sumber zat tambahan atau reagen yang beresiko Transmisible Spongiform Encephalophaties (TSE) / Bovine Spongiform Encephalophaties (BSE) n. Perubahan berat penyalut tablet atau berat cangkang kapsul pada bentuk sediaan oral immediate release o. Peningkatan, penambahan, penghilangan atau penggantian zat warna dan/atau pengaroma p. Pengurangan atau penghilangan satu atau lebih komponen dari zat warna dan/atau zat pengaroma Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 15 q. Perubahan atau penambahan imprint, bossing atau tanda lain (kecuali garis bagi) pada tablet atau printing pada kapsul, termasuk penggantian atau penambahan tinta yang digunakan untuk penandaan produk r. Perubahan sintesis zat tambahan (non Farmakope) s. Penggantian atau penambahan tempat pengemasan sekunder obat t. Pengetatan batas spesifikasi kemasan primer obat u. Perubahan komposisi secara kualitatif dan/atau kuantitatif dari bahan kemasan primer obat (untuk semua bentuk sediaan) v. Penambahan atau penggantian alat takar yang bukan merupakan bagian dari kemasan primer (tidak termasuk spacer device untuk metered dose inhaler) w. Perubahan minor pada prosedur analisis kemasan primer obat x. Perubahan prosedur pengujian bahan kemasan primer obat, termasuk penggantian atau penambahan prosedur pengujian y. Perubahan atau penambahan supplier komponen kemasan atau alat kesehatan yang menyertai obat, tidak termasuk supplier spacer devices untuk metered dose inhaler z. Pengurangan supplier komponen kemasan atau alat kesehatan yang menyertai obat , tidak termasuk supplier spacerdevices untuk metered dose inhaler aa. Penambahan parameter pengujian kemasan primer obat bb. Pengurangan batas kadaluarsa obat : Kemasan belum dibuka cc. Pengurangan batas kadaluarsa obat: Setelah kemasan dibuka atau setelah rekonstitusi 2. 6 Perubahan Penandaan Produk PT. Novartis Indonesia 2. 6. 1 Prosedur Perubahan Penandaan Produk PT. Novartis Indonesia Penandaan adalah keterangan yang lengkap mengenai khasiat, keamanan, cara penggunaannya serta informasi lain yang dianggap perlu yang dicantumkan pada etiket, brosur atau leaflet, dan kemasan primer serta sekunder yang disertakan pada obat (Kemenkes, 2008). Informasi minimal yang harus dicantumkan pada penandaan dapat dilihat pada Lampiran 3. Perubahan penandaan, baik yang terdapat pada leaflet maupun kemasan obat dapat disebabkan karena beberapa hal seperti adanya penambahan efek samping, Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 16 posologi, indikasi, perubahan kondisi penyimpanan, dan lain sebagainya. Berdasarkan jenis perubahan yang terjadi, perubahan yang berkaitan dengan penandaan ini dapat dikategorikan ke dalam registrasi variasi kategori 4 (empat), 5 (lima), ataupun 6 (enam) seperti yang telah dijelaskan sebelumnya (Badan POM RI, 2011). Perubahan ini sangat penting untuk diajukan ke Badan POM untuk menjamin keamanan obat saat digunakan oleh masyarakat. Alur perubahan penandaan yang terjadi pada produk-produk PT Novartis Indonesia diawali dengan adanya pemberitahuan terkait perubahan, baik yang terkait dengan mutu maupun keamanan oleh Novartis global atau pusat. Perubahan ini dapat bersifat urgent (3 hari kerja harus diajukan ke Badan POM), experdate (14 hari kerja harus diajukan ke Badan POM), atau standard (60 hari kerja harus diajukan ke Badan POM). Global memberitahukan adanya perubahan kepada DRA (Drug Regulatory Affairs)atau regulator melalui suatu sistem yang disebut DRAGON. DRAGON terbagi menjadi dua bagian, yaitu CDS (Core Data Sheet) dan CMC (Chemistry, Manufacturing, and Controls) terkait dengan jenis perubahan yang terjadi. Jika perubahan yang terjadi berkaitan dengan safety atau keamanan, maka perubahan ini dapat dilihat melalui CDS tracking list sedangkan untuk perubahan yang berkaitan dengan mutu, DRAGON akan memperlihatkannya melalui CMC tracking list. CDS tracking list dan CMC tracking list sendiri berisi link-link yang harus diunduh untuk memperoleh datadata perubahan yang terjadi secara lengkap (SOP of National Product Information, SOP No. DRA001, 2013). Proses selanjutnya, setelah mengunduh dan mengetahui perubahan apa yang terjadi, DRA mendaftarkan segera perubahan yang terjadi ke Badan POM. Terkait dengan perubahan penandaan, DRA akan menggunakan isi dari CDS sebagai dasar draft pertama dokumen registrasi yang akan diserahkan ke BPOM. DRA selanjutnya membuat tabel sandingan untuk membandingkan desain dan/ atau informasi produk yang telah disetujui versus desain dan/ atau informasi produk CDS saat ini serta mencantumkan penjelasan mengenai alasan terjadinya perubahan (disertai referensi pendukung jika ada). Dokumen yang akan diserahkan ke BPOM untuk registrasi ini harus mengikuti ketentuan Asean Common Technical Dossier (ACTD). Langkah selanjutnya dilakukan penyiapan Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 17 dossier oleh DRA (SOP of National Product Information, SOP No. DRA001, 2013). Setelah dossier disiapkan, maka selanjutnya dilakukan pemeriksaan terhadap dossier ini oleh DRA manager. Pada pemeriksaan ini, dilakukan pemastian akan akurasi dossier yang meliputi kelengkapan, kebenaran, dan konsistensi dossier berdasarkan checklist dokumen registrasi. Pada proses ini, orang yang bertanggung jawab dalam mempersiapkan dan memeriksa berkas adalah orang yang berbeda. Jika berkas tidak lolos verifikasi, DRA bertanggung jawab untuk menyiapkan ulang dan melengkapi berkas yang diperlukan. Setelah dossier diajukan ke BPOM, DRA bertanggung jawab dalam memasukkan tanggal penyerahan ke dalam "CDS tracking list"dan "Variation Node". Jika dossier dikembalikan atau ditunda oleh BPOM, maka DRA harus berkonsultasi terlebih dahulu dengan DRA Manager Global sebelum menanggapi hasil keputusan BPOM (SOP of National Product Information, SOP No. DRA001, 2013). Setelah menerima persetujuan dari Badan POM terkait dengan perubahan yang terjadi, maka DRA akan mengkomunikasikan informasi persetujuan kepada departemen terkait (yaitu Medical, Pemasaran, Penjualan, dan Technical Operations) dan membuat suatu formulir permintaan perubahan yang dikenal dengan istilah CRPM (Change Request of Printed Packaging Material) yang ditujukan kepada Packaging Development (SOP of National Product Information, SOP No. DRA001, 2013). Secara lebih rinci, alur dari mulai diberitahukannya adanya perubahan sampai dikeluarkannya CRPM dapat dilihat pada Gambar 2.2 dibawah ini. Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 18 [Sumber : SOP of National Product Information, SOP No. DRA001, 2013, telah diolah kembali] Gambar 2.2. Alur perubahan penandaan terkait safety update mulai dari pemberitahuan awal sampai pembuatan CRPM 2. 7 Produk B PT. Novartis Indonesia Produk B PT. Novartis Indonesia termasuk ke dalam kategori obat baru. Produk B diproduksi di Novartis Pharma Stein AG, Stein, Switzerland, sehingga termasuk ke dalam kategori obat impor. Produk B mengandung Indakaterol maleat sebagai zat aktif tunggal dengan bentuk sediaan berupa kapsul keras untuk Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 19 serbuk inhalasi. Produk ini terdiri dari 4 SKU (Stock Keeping Unit) dengan kekuatan dan besar kemasan yang berbeda, ditunjukkan pada Tabel 2.1. Tabel 2.1. SKU Produk B PT. Novartis Indonesia No. Bentuk Sediaan Kekuatan Jenis Kemasan Besar Kemasan 1 Kapsul + inhaler 150 mg Blister Dus, 1 blister @ 10 kapsul + 1 inhaler 2 Kapsul + inhaler 150 mg Blister Dus, 1 blister @ 30 kapsul + 1 inhaler 3 Kapsul + inhaler 300 mg Blister Dus, 1 blister @ 10 kapsul + 1 inhaler 4 Kapsul + inhaler 300 mg Blister Dus, 1 blister @ 30 kapsul + 1 inhaler Produk B mengandung Indakaterol maleat yang termasuk ke dalam obat bronkodilator dan antiasma yang diindikasikan untuk terapi pemeliharaan bronkodilator obstruksi aliran udara pada pasien dewasa dengan Penyakit Paru Obstruktif Kronik (PPOK). Indakaterol termasuk ke dalam “ultra” agonis beta2adrenergik dengan administrasi sekali sehari. Efek farmakologi dari beta2adrenergik yaitu stimulasi adenil siklase intraseluler (enzim yang mengkatalisis konversi adenosine triphosphate (ATP) menjadi cyclic-3’, 5’-adenosine monophosphate (cyclic monophosphate)). Peningkatan level siklik AMP menyebabkan relaksasi otot polos bronkial. Oleh karena itu, ketika dihirup indakaterol bekerja secara lokal pada paru-paru sebagai bronkodilator. Kontraindikasi dari produk B yaitu pasien dengan hipersensitifitas terhadap indakaterol atau salah satu zat tambahan dalam produk. Penggunaan produk B perlu diperhatikan pada penderita asma, hipersensitif, bronkospasme paradoksikal, hipokalemia, hiperglikemia, memburuknya penyakit, efek sistemik, dan efek kardiovaskuler. Efek samping yang umum dari produk ini pada dosis yang direkomendasikan yaitu nasofaringitis, infeksi saluran pernafasan atas, batuk, sakit kepala, dan kaku otot. Penggunaan produk B ini dengan rute inhalasi oral sekali sehari menggunakan inhaler yang terdapat dalam kemasan. Kapsul tidak boleh ditelan secara utuh. Dosis yang direkomendasikan yaitu inhalasi satu kali sehari yang mengandung 150 mg kapsul dengan menggunakan inhaler. Penggunaan dosis obat 300 mg sekali sehari menghasilkan keuntungan klinis pada pasien tertentu misalnya pada pasien PPOK yang parah. Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 20 BAB 3 METODOLOGI 3.1 Waktu Penyusunan dokumen registrasi dan proses pengajuan registrasi variasi dilaksanakan pada Praktek Kerja Profesi Apoteker (PKPA) periode 4 Februari 28 Maret 2014. 3.2 Lokasi Penyusunan dokumen registrasi dilakukan di PT. Novartis Indonesia, Jl. Prof. Dr. Satrio Kav. 18 Kuningan City Setiabudi Jakarta Selatan 12940, Indonesia. Sedangkan proses pengajuan registrasi variasi dilakukan di BPOM, Jl. Percetakan Negara No. 23 Jakarta 10560. 3.3 Data Data yang digunakan adalah dokumen terkait Produk B PT. Novartis Indonesia yang telah memiliki izin edar dan mengalami perubahan penandaan (safety update) baik bersifat major maupun minor disesuaikan dengan persyaratan dokumen yang harus diserahkan berdasarkan Buku Coklat 2011 (Peraturan Tata laksana registrasi obat diatur oleh Badan POM dalam keputusan Kepala Badan Pengawas Obat dan Makanan Republik Indonesia Nomor HK.03.1.23.10.11.08481 Tahun 2011 Tentang Kriteria dan Tata Laksana Registrasi Obat). 3.4 Metode Metode yang digunakan yaitu dengan menyusun dan mengajukan dokumen registrasi variasi Produk B PT. Novartis Indonesia dengan tahapan sebagai berikut: a. Mengumpulkan dan mempelajari informasi terkait perubahan produk dari Novartis global ke DRA Novartis Indonesia yang terdapat dalam sistem DRAGON. b. Melakukan pengambilan dokumen dari sistem informasi terkait perubahan penandaan pada produk dari sistem informasi REDI GO. Pengambilan 20 Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia 21 dokumen disesuaikan dengan persyaratan dokumen registrasi variasi dari BPOM, kemudian dokumen dicetak. c. Menyusun dossier sesuai dengan format ACTD. Selain menyusun dokumen yang diperoleh dari globat, juga dilakukan pembuatan surat atau dokumen lain yang diperlukan seperti membuat formulir registrasi, surat pengantar, mengisi checklist registrasi variasi, dan dokumen administratif lainnya. Pada tahap ini, dokumen dipelajari lebih lanjut untuk menentukan kategori registrasi yang ditulis dalam formulir registrasi. Kemudian, dilakukan pengecekan kelengkapan dokumen sebelum diajukan ke BPOM. d. Proses submit atau pengajuan dokumen di BPOM. Pada proses ini, terdapat proses evaluasi dokumen yang dilakukan oleh evaluator BPOM di loket registrasi, pengisian formulir elektronik registrasi (AeRO), dan juga penyerahan dokumen registrasi yang sudah lengkap ke loket penyerahan dokumen BPOM. Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 22 BAB 4 PEMBAHASAN Penandaan adalah keterangan yang lengkap mengenai khasiat, keamanan, cara penggunaannya serta informasi lain yang dianggap perlu yang dicantumkan pada etiket, brosur atau leaflet, dan kemasan primer serta sekunder yang disertakan pada obat (Kemenkes, 2008). Pada produk yang telah mendapatkan izin edar dapat terjadi perubahan, termasuk perubahan pada bagian penandaan yang terkait khasiat-keamanan produk. Perubahan penandaan, baik yang terdapat pada leaflet maupun kemasan obat dapat disebabkan karena beberapa hal seperti adanya penambahan efek samping, posologi, indikasi, perubahan kondisi penyimpanan, dan lain sebagainya. Segala jenis perubahan penandaan tersebut harus dilaporkan ke Badan Pengawas Obat dan Makanan (BPOM) melalui mekanisme Registrasi Variasi untuk menjamin keamanan obat saat digunakan oleh masyarakat. Berdasarkan jenis perubahan yang terjadi, perubahan yang berkaitan dengan penandaan ini dapat dikategorikan ke dalam registrasi variasi kategori 4 (variasi major), kategori 5 (variasi minor yang memerlukan persetujuan), ataupun kategori 6 (variasi minor dengan notifikasi) seperti yang telah dijelaskan sebelumnya (Badan POM RI, 2011). Pada Produk B PT. Novartis Indonesia, terdapat perubahan penandaan (safety update) yang diajukan pada bulan April 2014. Untuk melakukan perubahan tersebut, maka perlu dilakukan proses registrasi variasi terhadap produk tersebut. Pada proses registrasi variasi ini terdapat beberapa tahap yang dilakukan mulai dari adanya informasi perubahan produk dari global, hingga melakukan pengajuan atau submit dokumen ke BPOM yang prosesnya harus sesuai dengan Peraturan Tata laksana registrasi obat yang diatur oleh Badan POM dalam keputusan Kepala Badan Pengawas Obat dan Makanan Republik Indonesia Nomor HK.03.1.23.10.11.08481 Tahun 2011 Tentang Kriteria dan Tata Laksana Registrasi Obat. 22 Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia 23 4. 1 Informasi perubahan dari Novartis global ke DRA Novartis Indonesia. Alur perubahan penandaan yang terjadi pada produk B PT Novartis Indonesia diawali dengan adanya pemberitahuan mengenai perubahan, baik terkait dengan keamanan (safety) maupun mutu (quality) oleh Novartis global atau pusat ke bagian DRA (Drug Regulatory Affairs) atau regulator dari masing-masing negara dimana Produk B diedarkan atau dipasarkan. Global memberitahukan adanya perubahan kepada DRA melalui suatu sistem yang disebut DRAGON. DRAGON terbagi menjadi dua bagian, yaitu CDS (Core Data Sheet) dan CMC, terkait dengan jenis perubahan yang terjadi. Pada Produk B terjadi perubahan yang berkaitan dengan safety atau keamanan, maka Novartis global mengumumkan perubahan ini ke sistem DRAGON yang dapat dilihat melalui CDS tracking list kepada DRA di Indonesia dalam bentuk CDS. Perubahan pada Produk B terdapat pada CDS 30.09.13. Kemudian, DRA akan menilai apakah perubahan yang dilakukan perlu diajukan ke BPOM. Berdasarkan informasi dari DRAGON tersebut, pada produk B dilakukan perubahan penandaan yaitu pada bagian: a. Dosage and administration Penambahan informasi pada bagian “Metode administrasi” untuk membantu mengurangi potensi kesalahan penggunaan obat (seperti pasien sengaja menelan kapsul dibanding menggunakan perangkat inhalasi atau inhaler). b. Warning and precautions Bagian Asma : penambahan klim kelas label long-acting β2-drenergik receptor agonist (LABA) yang berpotensi meningkatkan resiko kematian terkait asma. Bagian Cardiovascular effects : penambahan klim potensi class effects yang berkaitan dengan LABA, juga mencakup “perpanjangan interval QT” dalam daftar potensi perubahan EKG. c. Adverse drug reactions Perubahan editorial dengan mengkoreksi jumlah N-nilai populasi keselamatan. d. Pharmacokinetics (PK) Perubahan editorial metabolit pada bagian metabolisme. Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 24 Setelah diketahui perubahan yang dilakukan, kemudian DRA menentukan apakah perubahan perlu diajukan ke BPOM atau tidak. Untuk perubahan Produk B seperti yang disebutkan sebelumnya, perlu dilakukan pengajuan registrasi variasi karena berkaitan dengan penandaan yang akan mempengaruhi aspek keamanan produk. 4. 2 Pengambilan dokumen dari sistem informasi terkait perubahan penandaan produk B. Setelah diketahui adanya perubahan penandaan yang dilakukan, kemudian dilakukan pengambilan dokumen dari sistem informasi CDS tracking list yang disebut REDI GO yang berisi link-link yang harus diunduh untuk memperoleh data-data perubahan pada penandaan secara lengkap. Pengambilan dokumen disesuaikan dengan persyaratan dokumen registrasi variasi dari BPOM. Dari REDI GO, didapatkan dokumen berupa leaflet yang akan diajukan, tabel sandingan atau rationale sheet dan beberapa referensi pendukung yang diperlukan, jika ada. Leaflet berisi keterangan produk secara lengkap terkait informasi produk berupa deskripsi dan komposisi, indikasi, posologi (dosis dan administrasi), kontraindikasi, perhatian dan peringatan, efek samping, interaksi, farmakologi klinis (mekanisme aksi, farmakodinamik, dan farmakokinetik), data keamanan non-klinis, kondisi penyimpanan, serta ukuran kemasan. Rational sheet berisi perbandingan antara informasi produk yang telah disetujui BPOM dengan informasi produk pada CDS yang diajukan disertai dengan alasan (rationale) yang berisi penjelasan mengenai alasan terjadinya perubahan, disertai referensi pendukung perubahan tersebut. Sedangkan referensi merupakan data-data pendukung dilakukannya perubahan, dapat berupa data klinis, data non-kinis, farmakope, jurnal ilmiah, buku acuan, dan referensi lain yang sifatnya ilmiah. Setelah mengunduh dokumen terkait perubahan dari sistem informasi REDI GO, kemudian dokumen dicetak dan dianalisis untuk menentukan kategori registrasi. Penentuan kategori registrasi variasi dilihat berdasarkan buku coklat. Berdasarkan perubahan yang dilakukan pada Produk B, kategorinya termasuk ke dalam kategori registrasi variasi Major (VaMa), yaitu Perubahan informasi produk atau penandaan yang mempengaruhi aspek khasiat-keamanan yang tidak Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 25 memerlukan data uji klinik. Perubahan pada bagian penandaan khususnya pada bagian posologi (dosage and administrasion) dan warning and precautions termasuk ke dalam kategori registrasi variasi major (VaMa), sedangkan perubahan editorial pada adverse drug reactions dan Pharmacokinetics termasuk ke dalam kategori registrasi variasi minor dengan notif (VaMi-A), yang keduanya samasama tidak memerlukan data uji klinik. Oleh karena itu, perubahan dikategorikan menjadi registrasi Variasi Major (VaMa). 4. 3 Penyusun dossier dan pengecekan kelengkapan dokumen Perubahan yang dilakukan pada Produk B termasuk ke dalam kategori Variasi Major, sehingga perlu dilakukan proses pra-registrasi sebelum proses registrasi untuk penentuan kategori registrasi, jalur evaluasi, biaya evaluasi, dan penentuan dokumen registrasi obat. Penyusunan dokumen dan proses pengajuan praregistrasi sesuai dengan persyaratan dokumen pra-registrasi yang terdapat pada buku coklat. Proses pra-registrasi ini sebelumnya telah dilakukan oleh staf DRA PT. Novartis Indonesia. Dari proses pra-registrasi, akan diperoleh Hasil PraRegistrasi (HPR) yang berisi identitas produk (nama obat, bentuk sediaan, zat aktif, kemasan, produsen dan NIE), kategori registrasi, jalur evaluasi, serta biaya evaluasi sesuai PP RI No. 48 tahun 2010 (Lampiran 4). Proses pengajuan registrasi dilakukan maksimal 1 tahun setelah HPR dikeluarkan. Oleh karena itu, langkah selanjutnya yaitu menyiapkan dokumen registrasi berupa dossier. Penyiapan dossier dilakukan dengan format dokumen mengikuti ketentuan Asean Common Technical Dossier (ACTD). Terkait dengan perubahan penandaan, isi dari CDS, rationale sheet, dan referensi yang mendukung akan digunakan sebagai dasar draft pertama dokumen registrasi yang akan diserahkan ke BPOM. Selain menyusun dokumen yang diperoleh dari global, terdapat beberapa kelengkapan yang perlu disiapkan ataupun dibuat, misalnya dokumen administrasi. Dokumen administrasi yang perlu dibuat diantaranya: a. Surat pengantar Surat pengantar untuk Produk B ditulis oleh pendaftar kepada Kepala Sub Direktorat Obat Baru Badan POM yang berisi permohonan pengajuan registrasi Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 26 variasi Produk B disertai dengan keterangan perubahan yang dilakukan dan dokumen penunjang pengajuan perubahan yang dilampirkan. b. Formulir registrasi tiap SKU, masing-masing rangkap 5. Format formulir registrasi dapat dilihat pada Lampiran 1. c. Pernyataan pendaftar Pernyataan pendaftar merupakan surat yang dibuat oleh pendaftar yang menyatakan bahwa semua informasi dalam dokumen registrasi untuk memperoleh persetujuan perubahan terhadap Produk B adalah benar. Selain itu, pendaftar juga menyatakan bahwa saya telah memeriksa dan bertanggung jawab atas semua dokumen yang diserahkan ke Badan POM. d. Surat pernyataan terkait pemenuhan persyaratan registrasi variasi Surat pernyataan terkait pemenuhan persyaratan registrasi variasi merupakan surat pengajuan registrasi variasi major terhadap Produk B yang berisi pernyataan oleh pendaftar bahwa tidak dilakukan perubahan apapun di luar perubahan yang diajukan ke Badan POM. Sedangkan, dokumen dokumen yang perlu dipersiapkan, khususnya untuk registrasi variasi Produk B sebagai produk impor yaitu: a. Izin industri farmasi PT. Novartis Indonesia. b. Sertifikat GMP Novartis Pharma Stein AG, Stein, Switzerland, termasuk data inspeksi terakhir. c. Hasil Pra-Registrasi (HPR). d. Fotokopi NIE beserta semua surat persetujuan perubahan Produk B yang telah diterbitkan Badan POM sebelumnya beserta lampirannya berupa leaflet pada persetujuan sebelumnya. Pada bagian depan ordner dibuat cover ordner dengan format yang telah ditentukan pada buku coklat seperti yang dapat dilihat pada Lampiran 5. Setelah dokumen-dokumen yang diperlukan siap dan telah dicetak, kemudian mengisi checklist registrasi variasi dari Badan POM sesuai dengan kategori registrasi variasi dan dokumen yang telah dikumpulkan (Lampiran 6). Selanjutnya, semua dokumen tersebut disusun menjadi dossier dalam ordner sesuai format ACTD berdasarkan urutan pada checklist registrasi agar tersusun rapi. Tiap dokumen Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 27 diberi pembatas dan pada bagian tepi dokumen diberi tanda agar lebih rapi dan mudah dalam mencari dokumen atau surat dalam dossier. Setelah dossier disiapkan, maka selanjutnya dilakukan pemeriksaan terhadap dossier oleh DRA manager. Pada pemeriksaan ini, dilakukan pemastian akan akurasi dossier yang meliputi kelengkapan, kebenaran, dan konsistensi dossier berdasarkan checklist dokumen registrasi. Pada proses ini, orang yang bertanggung jawab dalam mempersiapkan dan memeriksa berkas adalah orang yang berbeda. Jika, berkas tidak lolos verifikasi, bagian DRA bertanggung jawab untuk menyiapkan ulang dan melengkapi berkas yang diperlukan. Setelah dossier disiapkan, dokumen dipelajari lebih lanjut agar pada saat evaluasi pendaftar siap menghadapi dan dapat menjawab pertanyaan dari evaluator BPOM. 4. 4 Proses submit atau pengajuan dokumen di BPOM. Proses submit dokumen registrasi dimulai dengan pendaftaran registrasi Produk B secara online di www.antrianobat.com. Setelah mendaftar secara online, BPOM akan mengeluarkan jadwal untuk pelaksanaan registrasi produk tersebut yang akan diumumkan dan dikirimkan melalui email ke kantor industri farmasi pendaftar, berisi tanggal registrasi dan nomor antrian ke loket pengecekan dokumen oleh evaluator BPOM. Pendaftar akan melakukan proses submit dokumen sesuai dengan jadwal yang telah ditentukan tersebut. Proses submit atau pengajuan dokumen registrasi di Badan POM dapat dilihat pada Lampiran 7. Proses registrasi, dilakukan di BPOM, Jl. Percetakan Negara No. 23 Jakarta di Gedung pelayanan publik. Di Gedung Pelayanan Publik, terdapat tempat khusus untuk bagian registrasi produk obat dan produk biologi, yaitu di lantai 1 sebelah kanan. Terdapat sembilan loket di ruang registrasi Gedung pelayanan publik dan satu loket khusus untuk verifikasi SPB dan pengambilan kuota pengisian AeRO. Loket 1 untuk proses evaluasi pra-registrasi, loket 2 dan 3 untuk proses evaluasi registrasi (satu untuk loket registrasi sesuai antrian yang di email dan satu lagi untuk tambahan data registrasi). Loket 4, 5, dan 6 digunakan bila diperlukan proses desk consultation, yaitu proses evaluasi yang dilakukan antara evaluator dengan pendaftar sekaligus pemberian keputusan secara langsung terhadap proses evaluasi registrasi produk. Desk consultation dilakukan bila Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 28 terjadi penundaan atau penumpukan proses evaluasi dokumen di Badan POM, atau pada kondisi tertentu lainnya. Loket 7 atau loket A untuk loket penyerahan dokumen registrasi. Loket 8 atau loket B merupakan loket penyerahan dokumen pra-registrasi atau tambahan data. Terakhir, loket 9 atau loket C merupakan loket untuk surat keluar, misalnya pengambilan surat persertujuan registrasi dan pengambilan Nomor Izin Edar (NIE). Pertama, pendaftar mengantri di loket evaluasi registrasi (loket 2 atau 3) sesuai dengan nomor antrian registrasi variasi untuk produk B. Setelah nama PT. Novartis Indonesia dipanggil, pendaftar masuk ke loket evaluasi. Pada proses ini, dilakukan proses evaluasi dokumen yang dilakukan oleh evaluator BPOM di loket registrasi. Evaluasi meliputi pengecekan kesesuaian kategori registrasi dengan perubahan yang dilakukan, kesesuaian dokumen registrasi dengan persyaratan BPOM, kelengkapan dokumen, referensi yang dibutuhkan, dan proses tanya jawab terkait perubahan produk B. Pada proses ini, kemampuan komunikasi dan negosiasi regulator juga menentukan lolos tidaknya dokumen dalam proses evaluasi. Jika dokumen sudah lengkap dan benar, evaluator akan memberikan cap lengkap dan Surat Perintah Bayar (SPB) kepada pendaftar. Setelah memperoleh SPB, pendaftar harus membayar biaya registrasi sebesar yang telah tercantum di HPR, yaitu sebesar 8 juta rupiah untuk biaya evaluasi 4 SKU Produk B (tiap SKU 2 juta rupiah) ke bank yang telah ditunjuk oleh BPOM (Bank BNI). Pembayaran dilakukan dengan cara setor tunai dengan mencantumkan Badan POM RI pada bagian penerima. Pada bagian pengirim dicantumkan nama, alamat, dan nomor telepon industri farmasi pendaftar, yaitu PT.Novartis Indonesia, AXA TOWER lt. 26 Kuningan City Jl. Prof. Dr. Satrio Kav. 18 Kuningan – Setiabudi Jakarta 12940 Tel +62 21-304 80 600. Penyetoran dilakukan dengan memberikan keterangan tujuan pembayaran yaitu untuk praregistrasi atau registrasi serta mencantumkan nama produk. Contoh keterangan untuk penyetoran pembayaran registrasi produk B yaitu “Reg fee of Product B (safety update)”. Setelah dilakukan pembayaran, pendaftar akan mendapatkan bukti bayar dari bank yang bersangkutan. Bukti bayar yang diperoleh dari pembayaran biaya registrasi kemudian diserahkan ke loket pengambilan kuota pengisian AeRO. Kemudian pendaftar Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 29 mengisi formulir kuota pengisian AeRO (formulir elektronik registrasi di BPOM) di komputer yang telah disediakan. Selanjutnya pendaftar melakukan pengisian data-data Produk B untuk registrasi variasi di sistem AeRO tersebut. Lalu, dilakukan penyerahan dokumen registrasi yang sudah lengkap ke loket penyerahan dokumen registrasi Badan POM (loket 7). Setelah dokumen registrasi diserahkan, pendaftar akan mendapatkan nomor FERO yang merupakan nomor identitas registrasi produk untuk penelusuran proses registrasi. Selanjutnya dokumen akan mengalami proses evaluasi oleh Panitia Penilai sebagai evaluator sebelum Badan POM mengeluarkan surat persetujuan perubahan. Dokumen akan dievaluasi sesuai jalur registrasi variasi Produk B yaitu jalur 100 HK. Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 BAB 5 KESIMPULAN DAN SARAN 5.1 Kesimpulan Pada Produk B PT. Novartis Indonesia dilakukan registrasi variasi karena terdapat perubahan bagian penandaan (safety update) yang termasuk ke dalam kategori Variasi Major (VaMa) yaitu Perubahan informasi produk/penandaan yang mempengaruhi aspek khasiat-keamanan yang tidak memerlukan data uji klinik. Perubahan pada Produk B. Proses registrasi variasi dilakukan sebagai berikut: a. Informasi perubahan dari Novartis global ke DRA Novartis Indonesia b. Pengambilan dokumen dari sistem informasi terkait perubahan penandaan produk c. Penyusunan Dossier dan pengecekan kelengkapan dokumen d. Proses submit atau pengajuan dokumen di BPOM Dokumen yang diserahkan ke BPOM untuk registrasi variasi mengikuti ketentuan ACTD dan penyusunan serta pengajuannya berdasarkan pada Peraturan Tata laksana registrasi obat yang diatur oleh BPOM dalam keputusan Kepala Badan Pengawas Obat dan Makanan Republik Indonesia Nomor HK.03.1.23.10.11.08481 Tahun 2011 Tentang Kriteria dan Tata Laksana Registrasi Obat. 5.2 Saran a. Lebih banyak kerjasama antara Industri Farmasi (PT. Novartis Indonesia) bagian DRA dengan Program Studi Apoteker dalam pelaksanaan PKPA untuk membekali para calon apoteker dengan pengetahuan dan keterampilan dalam proses registrasi. b. Perlu diadakan pelatihan secara berkala mengenai registrasi kepada staf bagian DRA agar dapat terus mengembangkan pengetahuan dan kemampuannya di bidang registrasi. c. Perlu dibuatnya SOP (Standar Operational Procedure) terkait kegiatan yang berkaitan dengan registrasi yang dilakukan oleh bagian DRA mulai dari penerimaan informasi perubahan produk di sistem DRAGON, penyusunan dokumen, pengajuan registrasi, penerimaan approval BPOM hingga penyimpanan dokumen registrasi agar lebih terarah. 30 Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia DAFTAR PUSTAKA Badan POM RI. (2011). Peraturan Kepala Badan Pengawas Obat dan Makanan Nomor HK.03.1.23.10.11.08481 Tahun 2011 tentang Kriteria dan Tata Laksana Registrasi Obat. Jakarta: Badan POM RI. Fauzia. (2013). Review Implementasi Persetujuan BPOM terhadap Current Product PT. Novartis Indonesia yang terdiri dari Komponen Produk Lokal, Leaflet Produk yang Diambil Secara Acak, serta Komponen Produk Untuk Bulan Agustus. Depok : Universitas Indonesia, 10-13. Kementerian Kesehatan. (2008). Peraturan Menteri Kesehatan No. 1120/ MENKES/ PER/ XI/ 2008 Tentang Perubahan Atas Peraturan Menteri Kesehatan Nomor 1010/ MENKES/ PER/ XI/ 2008 Tentang Registrasi Obat. Jakarta: Kementerian Kesehatan Republik Indonesia. Kementerian Kesehatan. (2008). Peraturan Menteri Kesehatan No. 1010/ MENKES/ PER/ XI/ 2008 Tentang Registrasi Obat. Jakarta: Kementerian Kesehatan Republik Indonesia. Novartis. (2014). Package leaflet: Information for the user. Mei 10, 2014. http://www.novartis.com.au/DownloadFile.aspx?t=c&f=exe.pdf&dateid=12949 31209000 Novartis Indonesia. (2014). Tentang Novartis. http://www.id.novartis.com. Diakses tanggal Maret 8, 2014. Standard Operating Procedure of National Product Information, SOP No. DRA001, Effective Date: 31-May-2013. The Asean Common Technical Dossier (ACTD) For The Registration of Pharmaceuticals For Human Use - Organization Of The Dossier 31 Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 Universitas Indonesia LAMPIRAN Universitas Indonesia Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 33 Lampiran 1. Formulir registrasi obat dan produk biologi Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 34 Lampiran 1. (Lanjutan) Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 35 Lampiran 1. (Lanjutan) Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 36 Lampiran 1. (Lanjutan ) Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 37 Lampiran 2. Dokumen administratif registrasi variasi Dokumen administratif yang harus diserahkan pada saat pengajuan registrasi variasi meliputi: 1. Surat Penngantar. 2. Formulir Registrasi. 3. Pernyataan Pendaftar. 4. Sertifikat dan Dokumen Administratif (sesuai dengan status produksi: obatproduksi dalam negeri, kontrak, lisensi, ekspor, impor) sesuai Lampiran 5. 5. Hasil Pra Registrasi (jika dipersyaratkan). 6. Kuitansi/Bukti Pembayaran. 7. Dokumen lain-lain. 7.1.Surat pernyataan terkait pemenuhan persyaratan registrasi variasi (misal: surat pernyataan bahwa prosedur pengujian zat aktif tidak berubah untuk registrasi variasi pengetatan batas spesifikasi zat aktif). 7.2.Copy NIE dan semua surat persetujuan perubahan yang diterbitkan oleh Badan POM beserta lampirannya. 7.3.Tabel sandingan perubahan yang diajukan, termasuk referensi perubahan. 7.4. Justifikasi terhadap perubahan yang diajukan. [Sumber : Anonim, 2011] Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 38 Lampiran 3. Informasi Minimal yang Harus Dicantumkan Pada Penandaan No. Informasi yang harus dicantumkan Bungkus Luar Catch Cover / Amplop Etiket/ Blister/ Strip label Etiket Ampul / Vial 1. Nama Obat √ √ √ √ √ 2. Bentuk Sediaan √ √ √ (-) √ d) 3. Besar kemasan (unit) √ √ √ (-) √ 4. Nama dan kekuatan zat aktif √ √ √ √ √ 5. Nama dan alamat pendaftar √ √ √ √ c) √ c) 6. Nama dan alamat produsen √ √ √ √ c) √ e) 7. Nama dan alamat pemberi lisensi √ √ √ √ c) (-) 8. Cara pemberian √ √ √ (-) √ 9. Nomor izin edar √ √ √ √ √ 10 Nomor bets √ √ √ √ √ 11. Tanggal produksi √ √ (-) (-) (-) 12. Batas kadaluarsa √ √ √ √ √ 13. Indikasi √ a) √ √ b) (-) (-) 14. Posologi √ a) √ √ b) (-) (-) 15. Kontraindikasi √ b) √ √ b) (-) (-) 16. Efek samping √ b) √ √ b) (-) (-) 17. Interaksi obat √ b) √ √ b) (-) (-) 18. Peringatan-perhatian √ √ √ (-) (-) b) Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 b) 39 Lampiran 3. (Lanjutan) No. 19. 20. 21. Informasi yang harus dicantumkan Peringatan khusus, misalnya : a. “Harus dengan resep dokter" Bungkus Luar Catch Etiket/ Blister/ Etiket Cover / Label Strip Ampul / Amplop Vial √ √ √ √ √ b. Tanda peringatan (P. No. 1 – P. No. 6) √ √ √ (-) (-) c. Kotak peringatan √ √ √ (-) (-) d. "Bersumber babi/bersinggungan" √ √ √ (-) √ e. Kandungan alkohol √ √ √ (-) √ Cara penyimpanan obat (termasuk cara penyimpanan setelah rekonstitusi) Penandaan khusus, misalnya: √ √ √ (-) (-) a. Harga eceran tertinggi (HET) √ √ √ √ b. Logo golongan obat (obat keras/ bebas terbatas/bebas) √ √ √ (-) √ d) (-) c. Logo generik (khusus untuk obat generik) √ √ √ √ √ d) Keterangan: a) Harus dicantumkan untuk obat bebas dan obat bebas terbatas, untuk obat keras dapat merujuk pada informasi produk untuk pasien. b) Informasi dapat merujuk pada informasi produk untuk pasien. c) Dicantumkan nama pendaftar atau nama produsen atau nama pemberi lisensi. d) Dikecualikan untuk ampul atau vial kurang dari 10 mL. e) Hanya nama negara [Sumber : Anonim, 2011] Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 40 Lampiran 4. Hasil Pra-Registrasi (HPR) Produk B Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 41 Lampiran 5. Cover ordner Produk B Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 42 Lampiran 6. Contoh Checklist registrasi Variasi Major (VaMa) Produk B dan cara pengisiannya. Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 43 Lampiran 6. (Lanjutan) Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 44 Lampiran 6. Lanjutan Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014 45 Lampiran 7. Proses submit atau pengajuan dokumen registrasi variasi di Badan POM Laporan praktik…, Asvinastuti Rikasih, FFar UI, 2014